
![]()
Peutz-Jeghers syndrome
Peutz-Jeghers syndrome (PJS) is a rare autosomal dominant disorder characterised by the development of mucocutaneous lentiginosis and multiple hamartomatous polyps throughout the gastrointestinal tract, particularly the small intestine. It is caused primarily by pathogenic variants in the STK11/LKB1 gene, a tumour suppressor located on chromosome 19p13.3.
Clinically, patients often present in childhood with bluish-black pigmented macules on the lips, oral mucosa, fingers, and perianal region. Gastrointestinal manifestations include abdominal pain, intussusception, gastrointestinal bleeding, and obstruction secondary to polyps. Polyps are histologically distinct, featuring arborising smooth muscle cores, and typically do not follow the adenoma-carcinoma sequence seen in other polyposis syndromes.
PJS significantly increases lifetime risk for a range of malignancies, including colorectal, gastric, pancreatic, breast, ovarian, cervical, and testicular cancers. Surveillance and early polypectomy are central to management, with guidelines recommending lifelong endoscopic and oncologic monitoring.
Though initial observations trace back to the 19th century, the condition was fully delineated as a clinical syndrome in the mid-20th century. The eponym honours Jan Peutz, who first described familial polyposis with pigmentation in 1921, and Harold Jeghers, who synthesised historical and clinical evidence into a cohesive syndrome in 1949.
Historical timeline of Peutz-Jeghers syndrome
1895 – J. T. Conner published the first cases of two 9-year old twins with small pigmented spots scattered over the lips, gums and hard palate, but not on the tongue. These twins were described to be of dark complexion and anaemic.
Dr. J. T. CONNER showed two cases of Pigmentation of the Lips and Mouth in twins, both girls, aged twelve years, of dark complexion and anæmic. The pigment spots, which were only noticed two years ago, were of ink-black colour, mostly of very small size, and scattered over the lips (especially the lower), gums, and hard palate, but not on the tongue
1896 – Sir Jonathan Hutchinson published the portraits and further description of the twins described by Connor in his article titled ‘Pigmentation of lip and mouth‘. He wrote that these twins “developed a number of black pigmented spots on the lips and inside of the mouth“, which had increased in size and number since appearing at three-years of age. Sir Hutchinson considered the pigmented spots were not pathological due to the good health of the subjects.
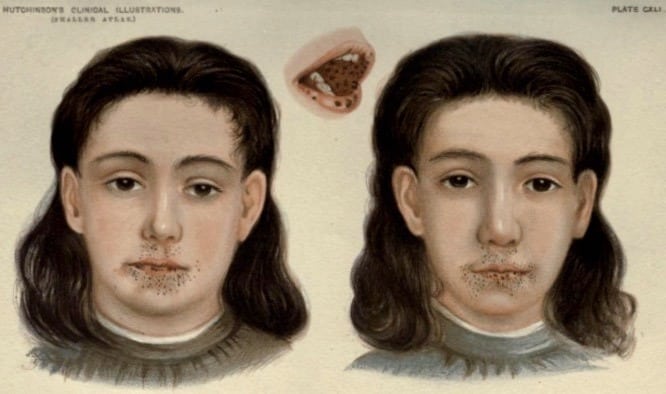
1919 – Frederick Parkes Weber (1863-1972) published an update on these twins, confirming that one of the twins died following a surgical procedure for intussusception at age 20-years. Intestinal polyposis was not specifically confirmed as the cause of intussusception. The second twin died at age 52-years of breast cancer, confirmed by her brother to Jeghers et al.
1921 – Johannes Laurentius Augustinus Peutz (1886-1957) described a 15-year-old boy with poor appetite, and intermittent abdominal pain, later developing an ileus from jejunal polyps. The patient had numerous distinctive pigmented spots on his face and mouth, with numerous ‘pea to grape sized’ rectal polyps and rectal pigmentation also observed on rectoscopy. Evaluation of the patient’s family revealed similar pigmentation in the father, and 5 of the 7 siblings. The father had two sisters who died from intestinal obstruction.
Peutz considered that this was a congenital and familial syndrome.
Original
English
Terwijl nu het voorkomen der polypen in den darm als een familiaire ziekte een welbekend iets is, geven mijn ge vallen een dusdanige aanvulling van de kennis van het ziektebeeld, waarover ik tenminste in de literatuur nog niets vond vermeld. Want ik vond nl., zooals reeds werd gezegd, behalve de darmpolypen nog in 2 der 5 gevallen neuspolypen en van de 7 kinderen 5 met de beschreven pigmentatie benevens de 2 gevallen – zusters van den vader – waar anamnestisch ook diezelfde pigmentatie aan wezig bleek. Het lijkt mij zeer waarschijnlijk dat ook deze veranderingen in samenhang staan met de congenitale stoor nis, die tot het optreden van de polyposis geleid heeft.
While the occurrence of the polyps in the intestine as a familial disease is well-known, my cases add to the knowledge of the clinical picture, not previously reported in the literature. In addition to the intestinal polyps I found nasal polyps (2 of the 5 cases) and the described mucous membrane pigmentation (5 of the 7 children). In a further 2 cases – sisters of the father – the same pigmentation was present. It seems to me very likely that these changes are related to the congenital disorder which led to the appearance of the polyposis.
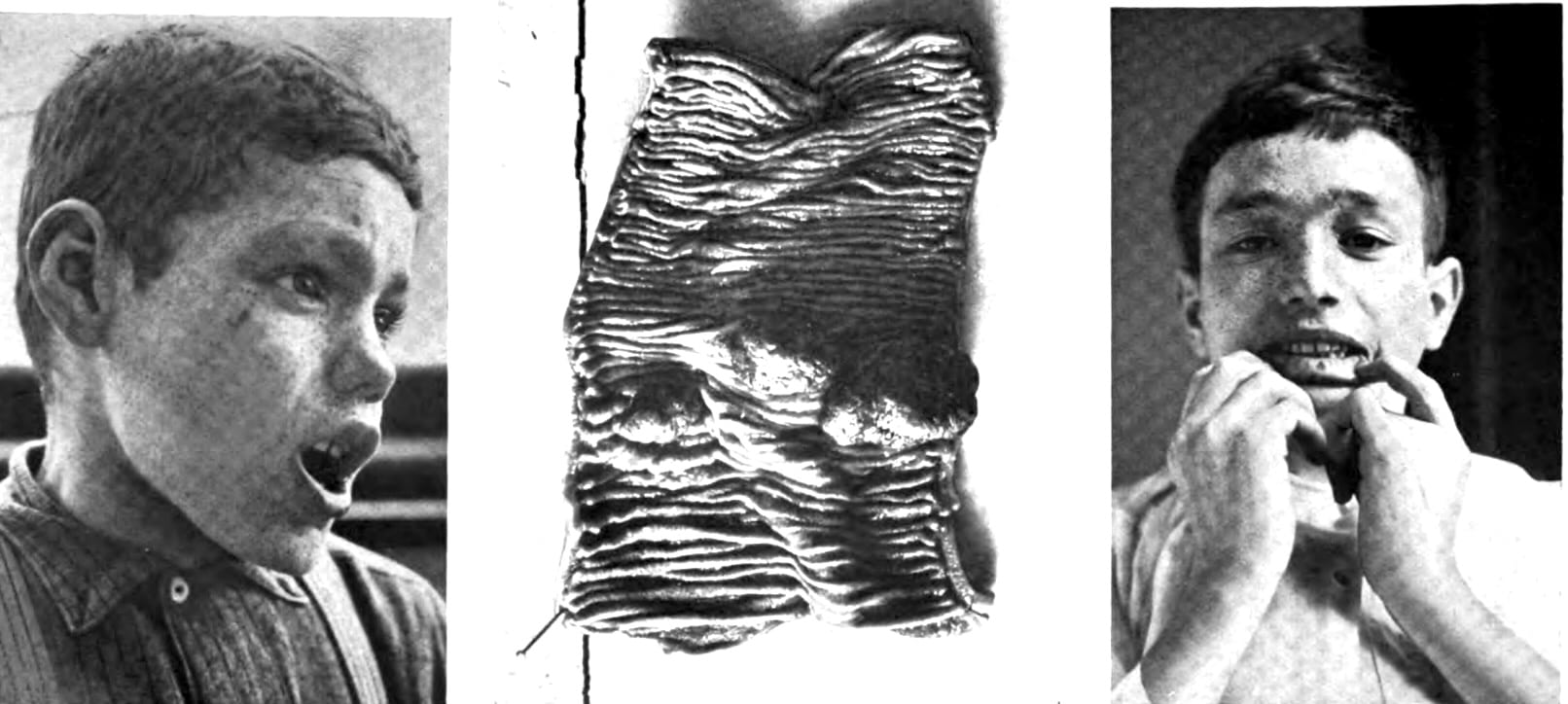
Peutz 1921
1944 – Harold Joseph Jeghers (1904-1990) published two cases in his three-part NEJM review article titled ‘Pigmentation of the Skin‘. He provided a brief literature review of two similar cases with skin pigmentation and polyposis reported by Hutchinson in 1896 (later confirmed to have intussusception by Weber in 1919), and Metzger et al. (1935)
Case 1
Case 2
A 14-year old American schoolgirl who presented to the Fifth Medical Service of Boston City Hospital with 6 weeks of persistent watery non-bloody diarrhoea 5-6 times per day. Her ileum was resected in 1933 following the second episode of intussusception and intestinal obstruction, which found polyps of the stomach, ileum and sigmoid. The patient had numerous small, brown-black spots around the mouth, lips and oral mucous membrane, in addition to the dorsal fingers and toes. The spots on the lips were present since early childhood according to the parents.
A 39-year old Italian-French woman who presented with a protruding rectal mass, and 9 months of constipation and intermittent rectal bleeding. She had previously been admitted with abdominal pain, nausea and vomiting. Numerous brown and bluish-brown pigmented spots on the face, eyes, mouth, lips, gums, fingers and toes were noted. Multiple small and large intestine polyps were seen on gastrointestinal x-rays. Ileostomy, partial colectomy, fulguration of polyps in the rectosigmoid and anastomosis between the ileum and rectosigmoid were performed; however, the patient subsequently died of intraperitoneal and wound infection. Autopsy demonstrated polyps in the stomach, duodenum, jejunum, ileum, and rectum, with a brown-gray ‘fish-skin’ appearing small intestine mucosa.
1949 – Jeghers, Victor Almon McKusick (1921-2008) and Kermit Harry Katz (1914-2003) established the combination of polyposis and pigmentation of the skin and mucous membranes as a distinct clinical syndrome internationally in their case series of 10 patients and literature review. They predicted that the pigmented spots and polyps were caused by mutations in a single gene.

1954 – The eponymous term ‘Peutz-Jeghers syndrome’ was introduced by the radiologist André Bruwer (1918-2008) in his paper ‘Surface pigmentation and generalized intestinal polyposis; (Peutz-Jeghers syndrome)‘
1998 – Hemminki et al. and Jenne et al. independently identified germline mutations in STK11/LKB1 on chromosome 19p encoding for the tumour suppressor serine/threonine kinase in patients with Peutz-Jeghers syndrome.
1999 – Westerman et al. published an extensive follow-up of six-generations of the original family described in 1921 by Peutz. Gastrointestinal polyposis, mucocutaneous pigmentation, nasal polyposis, and rectal extrusion of polyps were prominent in the family. Intestinal obstruction and the development of malignant disease contributed significantly to mortality. DNA-mutation analysis revealed an inactivating mutation in the LKB1 gene, co-segregating with the original family’s disease phenotype.
2000–2020s – Multiple studies clarified STK11’s tumour suppressor role, genetic heterogeneity, and associated cancer risks; surveillance guidelines were refined.
Associated Persons
- Sir Jonathan Hutchinson (1828-1913)
- Johannes Laurentius Augustinus Peutz (1886-1957)
- Harold Joseph Jeghers (1904-1990)
Alternative names
- Jeghers syndrome; Jeghers-Peutz syndrome
- Peutz disease
References
Original articles
- Conner JT. Aesculapian Society of London. Lancet. 1895;2:1169.
- Hutchinson J. Pigmentation of lip and mouth. Archives of Surgery, 1896; 7: 290. [Plate CXLI]
- Peutz JLA. Over een zeer merkwaardige, gecombineerde familiaire polyposis van de slijmvliezen, van den tractus intestinalis met die van de neuskeelholte en gepaard met eigenaardige pigmentaties van huid- en slijmvliezen. Nederlandsch Maandschrift voor Geneeskunde. 1921; 10: 134-146 [PDF]
- Jeghers H, McKusick VA, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med. 1949;241(25): 993-1005.
- Jeghers H, McKusick VA, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med. 1949;241(26):1031-1036.
- Weber FP. Patches of deep pigmentation of the oral mucous membrane not connected with Addison’s disease. Quarterly Journal of Medicine, Oxford, 1919; 12: 404-408.
Review articles
- Van Wijk ThW. Over het syndroom polyposis adenomatosa gastrointestinalis generalisatia heredofamiliaris gecombineerd met huid- en slijmvliespigementaties of ziekte van Peutz (thesis). 1950. Leiden.
- Bruwer A., Bargen JA. Kierland RR. Surface pigmentation and generalized intestinal polyposis; (Peutz-Jeghers syndrome). Proceedings of the Staff Meetings Mayo Clinic. 1954; 29(6): 168–171.
- Westerman AM, Entius MM, de Baar E, et al. Peutz-Jeghers syndrome: 78-year follow-up of the original family. Lancet. 1999;353(9160):1211-1215.
- Keller JJ, Offerhaus GJ, Giardiello FM, Menko FH. Jan Peutz, Harold Jeghers and a remarkable combination of polyposis and pigmentation of the skin and mucous membranes. Fam Cancer. 2001;1(3-4):181-185.
- Amru RL, Dhok A. Peutz-Jeghers Syndrome: A Comprehensive Review of Genetics, Clinical Features, and Management Approaches. Cureus. 2024 Apr 24;16(4):e58887
eponymictionary
the names behind the name
Doctor in Australia. Keen interest in internal medicine, medical education, and medical history.
BA MA (Oxon) MBChB (Edin) FACEM FFSEM. Emergency physician, Sir Charles Gairdner Hospital. Passion for rugby; medical history; medical education; and asynchronous learning #FOAMed evangelist. Co-founder and CTO of Life in the Fast lane | On Call: Principles and Protocol 4e| Eponyms | Books |