
![]()
Gitelman Syndrome
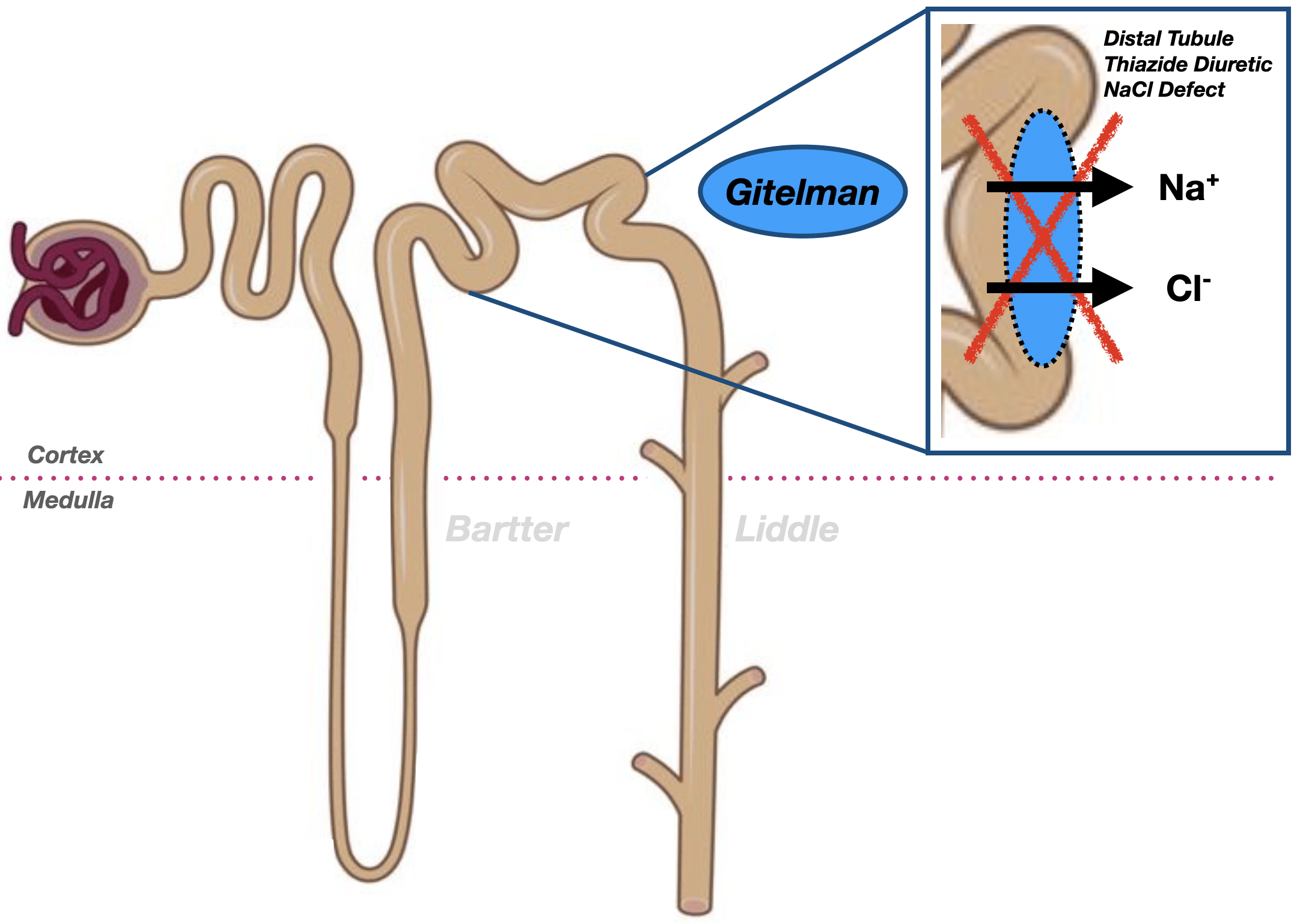
Gitelman syndrome affects the distal convoluted tubule, where a defect in the Na⁺-Cl⁻ cotransporter (SLC12A3) leads to hypokalaemia, metabolic alkalosis, hypomagnesaemia, and hypocalciuria, mimicking the action of thiazide diuretics.
Gitelman syndrome is an autosomal recessive renal tubulopathy characterised by hypokalaemia, hypomagnesaemia, hypocalciuria, and metabolic alkalosis, with normal or low blood pressure. It affects the distal convoluted tubule, mimicking the action of thiazide diuretics. First described by Hillel Gitelman in 1966 and genetically defined in the 1990s, it is now recognised as one of the most common inherited salt-wasting disorders.
Molecular Genetics and Diagnostic Criteria
In the mid-1990s, Gitelman syndrome was linked to mutations in the SLC12A3 gene encoding the thiazide-sensitive Na⁺-Cl⁻ cotransporter (NCC) in the distal convoluted tubule. This clarified its pathophysiology and confirmed it as a discrete entity from Bartter syndrome, which involves NKCC2 or ROMK mutations in the loop of Henle.
Diagnosis:
- Persistent hypokalaemia and hypomagnesaemia
- Hypocalciuria (vs. hypercalciuria in Bartter)
- Normal or low blood pressure
- No response to spironolactone
- Genetic confirmation of SLC12A3 mutations
Management:
- Consideration of amiloride, spironolactone or NSAIDs in select cases
- Lifelong potassium and magnesium supplementation; amiloride, spironolactone, and NSAIDs may assist in some cases.
- Cardiac monitoring is advised in symptomatic patients.
- Pharmacological chaperones (e.g., 4-phenylbutyrate) are being explored to stabilise mutant NCC protein trafficking.
History
1966 – Original description by Hillel Jonathan Gitelman (1932-2015), Graham JB, and Welt LG. published as A familial disorder characterized by hypokalemia and hypomagnesemia. Gitelman’s team documented clinical and biochemical features distinguishing the disorder from Bartter syndrome. Their patients had low urinary calcium and no juxtaglomerular hyperplasia, setting the stage for recognition of a distinct renal salt-handling defect.
The syndrome presented in two sisters revealed consistent electrolyte abnormalities — notably hypokalaemia and hypomagnesaemia — without significant renal sodium wasting or hypertension.
Gitelman, 1966
Comparative Context:
Comparison of Bartter, Gitelman and Liddle syndromes
| Feature | Bartter Syndrome | Gitelman Syndrome | Liddle Syndrome |
|---|---|---|---|
| Defect location | Thick Ascending Limb of Loop of Henle | Distal Convoluted Tubule | Collecting Duct (ENaC channel) |
| Transporter affected | NKCC2 (Na⁺-K⁺-2Cl⁻ cotransporter) | Na⁺-Cl⁻ cotransporter (SLC12A3) | Epithelial Na⁺ Channel (ENaC; SCNN1B/SCNN1G) |
| Pathophysiologic mimic | Loop diuretics (lose Ca²⁺) | Thiazide diuretics (preserve Ca²⁺) | Aldosterone excess (but low aldosterone) |
| Serum potassium (K⁺) | ↓ Hypokalaemia | ↓ Hypokalaemia | ↓ Hypokalaemia |
| Serum bicarbonate (HCO₃⁻) | ↑ Metabolic alkalosis | ↑ Metabolic alkalosis | ↑ Metabolic alkalosis |
| Serum magnesium (Mg²⁺) | Normal or mildly ↓ | ↓ Hypomagnesemia | Normal |
| Urinary calcium | ↑ Hypercalciuria | ↓ Hypocalciuria | Normal |
| Blood pressure | Normal or low | Normal or low | ↑ Hypertension |
| Renin | ↑ Elevated | ↑ Elevated | ↓ Suppressed |
| Aldosterone | ↑ Elevated | ↑ Elevated | ↓ Suppressed |
| Age of onset | Neonatal/Childhood | Childhood/Adolescence | Childhood/Adolescence |
| Response to treatment | NSAIDs (↓ prostaglandins), K⁺, spironolactone | Mg²⁺ and K⁺ supplementation, ± NSAIDs | Amiloride or triamterene (ENaC inhibitors) |
Associated Persons
- Frederic Crosby Bartter (1914-1983) | Bartter Syndrome
- Grant Winder Liddle (1921-1989) | Liddle Syndrome
- Hillel Jonathan Gitelman (1932-2015) | Gitelman Syndrome
References
Historical references
- Liddle GW, Bledsoe T, Coppage WS. A familial renal disorder stimulating primary aldosteronism, but with negligible aldosterone secretion. Trans Assoc Am Phys 1963; 76: 199-213
Eponymous term review
- Botero-Velez M, Curtis JJ, Warnock DG. Brief report: Liddle’s syndrome revisited–a disorder of sodium reabsorption in the distal tubule. N Engl J Med. 1994 Jan 20;330(3):178-81.
- Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, Lifton RP. Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995 Sep;11(1):76-82.
- Awadalla M, Patwardhan M, Alsamsam A, Imran N. Management of Liddle Syndrome in Pregnancy: A Case Report and Literature Review. Case Rep Obstet Gynecol. 2017;2017:6279460.
- Enslow BT, Stockand JD, Berman JM. Liddle’s syndrome mechanisms, diagnosis and management. Integr Blood Press Control. 2019 Sep 3;12:13-22.
eponymictionary
the names behind the name

BMedSci (Pharm) MB ChB, Edinburgh University. Emergency and Internal Medicine training. Interested in neuropharmacology and electrophysiology
BA MA (Oxon) MBChB (Edin) FACEM FFSEM. Emergency physician, Sir Charles Gairdner Hospital. Passion for rugby; medical history; medical education; and asynchronous learning #FOAMed evangelist. Co-founder and CTO of Life in the Fast lane | On Call: Principles and Protocol 4e| Eponyms | Books |