
![]()
Frederick Weber

Frederick Parkes Weber (1863-1962) was an English physician and dermatologist
His clinical writings span more than 1,200 articles and 23 books, establishing him as one of the most productive medical authors of his era. Weber’s keen diagnostic insight contributed to the characterisation of vascular anomalies and rare dermatological conditions, including Rendu-Osler-Weber disease and Sturge-Weber-Kalischer syndrome.
Educated at Charterhouse School, Trinity College Cambridge, and St. Bartholomew’s Hospital, Weber augmented his training in Vienna and Paris. He served as physician to the German Hospital, Dalston, for nearly five decades and also held posts at Mount Vernon Hospital and Brompton Hospital. His dedication to medicine extended well into his later years, remaining active in clinical practice and the Royal Society of Medicine into his 90s.
Weber was also a scholar of medical philosophy and the arts. His book Aspects of death and correlated aspects of life in art, epigram, and poetry reflected a unique blend of scientific inquiry and humanistic perspective. Beyond medicine, he was an accomplished numismatist, collaborating with his father Sir Hermann Weber (1823-1918) to build a renowned coin collection now housed in major museums worldwide.
Biography
- Born May 8, 1863 in London; eldest son of Sir Hermann David Weber (1823–1918), personal physician to Queen Victoria, and Matilda Gruning.
- 1874–1881 – Educated at Temple Grove School, East Sheen; then Charterhouse School.
- 1882 – Entered Trinity College, Cambridge.
- 1886 – BA, Cambridge.
- 1889 – MB BCh, Cambridge.
- 1890 – MA, Cambridge; MRCP.
- 1892 – MD, Cambridge; completed doctoral thesis on nephritis and pulmonary tuberculosis.
- 1890s – House Surgeon and House Physician at St. Bartholomew’s Hospital; House Physician at Brompton Hospital; postgraduate studies in Vienna and Paris.
- 1894 – Honorary Physician to German Hospital, Dalston; began tenure lasting nearly 50 years.
- 1894 – Joined Pathological Society of London; frequent contributor to its Transactions and Journal.
- 1899–1911 – First Assistant Physician, then Physician, at Mount Vernon Hospital for Chest Diseases.
- 1906 – Disposed of personal coin collection; significant donations made to British Museum, Bodleian Library, Fitzwilliam Museum, and Boston Medical Library.
- 1921 – Delivered first Mitchell Lecture at Royal College of Physicians: The Relations of Tuberculosis to General Bodily Conditions and to Other Diseases.
- 1930 – Awarded Moxon Gold Medal by the Royal College of Physicians for distinguished clinical observation and research.
- 1943 – Festschrift presented on his 80th birthday: seven volumes of reprints and bibliography of nearly 1,000 works.
- Died June 2, 1962 in his 100th year; survived by his wife Dr Hedwig Unger-Laissle.
Medical Eponyms
Klippel-Trénaunay-Weber syndrome (1900)
Klippel-Trenaunay syndrome (KTS) is a rare congenital cutaneous vascular malformation syndrome. Diagnosis is made with two of the three classic signs of localised cutaneous capillary malformations, venous abnormalities, and limb hypertrophy.
Also known as capillary-lymphatic-venous malformation (CLVM). It is associated with a a wide spectrum of clinical findings that can manifest during infancy and can progress throughout childhood and adulthood.
This syndrome is part of the PIK3CA-related overgrowth spectrum of diseases, which are caused by mutations in the PIK3CA gene.
1900 – Maurice Klippel (1858-1942) and Paul Trénaunay (1875-1938) reported a patient with asymmetrical hypertrophy of the soft tissue and bone, together with haemangiomatous lesions of the skin, using the term “naevus variqueux ostéo-hypertrophique”
1907 – Weber described three more cases and proposed the classic triad of “dermal naevi, osseous and soft tissue hemihypertrophy, and varicose veins.“
1918 – Weber revised the description to “hemangiectatic hypertrophy of limbs, congenital phlebarteriectasis and so-called congenital varicose veins“. He added an additional component, congenital arteriovenous fistula. Thereafter the triple-barreled eponym came into use.
Peutz-Jeghers syndrome
1896 – Sir Jonathan Hutchinson published the portraits and further description of the twins described by Connor in his article titled ‘Pigmentation of lip and mouth‘. He wrote that these twins “developed a number of black pigmented spots on the lips and inside of the mouth“, which had increased in size and number since appearing at three-years of age
1919 – Weber published an update on the twins, confirming that one of the twins died following a surgical procedure for intussusception at age 20-years. Intestinal polyposis was not specifically confirmed as the cause of intussusception. The second twin died at age 52-years of breast cancer, confirmed by her brother to Harold Joseph Jeghers (1904-1990)
Pfeifer-Weber-Christian disease (PWCD)
Rare idiopathic disease characterised by lobular panniculitis of adipose tissue with systemic symptoms and multiple organ involvement. PWCD is a rare illness of unknown aetiology, with a higher prevalence in females.
It is characterised by recurrent fever associated with the appearance of single or multiple non-suppurative nodules, sometimes tender, ranging from 1-12cm in diameter. The trunk and extremities, particularly the thighs and legs, are most frequently affected. Hands, face, and feet usually spared.
1892 – German physician, Victor Pfeifer (1846-1921) was the first to first describe multiple areas of atrophy in subcutaneous fatty tissue
1925 – Weber presented a case of a woman he treated at the German hospital in 1924 and termed the process relapsing non-suppurative nodular panniculitis.
1928 – The American physician, Henry Asbury Christian (1876-1951) highlighted the relapsing nature of the relapsing fever associated with the nodular nonsuppurative panniculitis
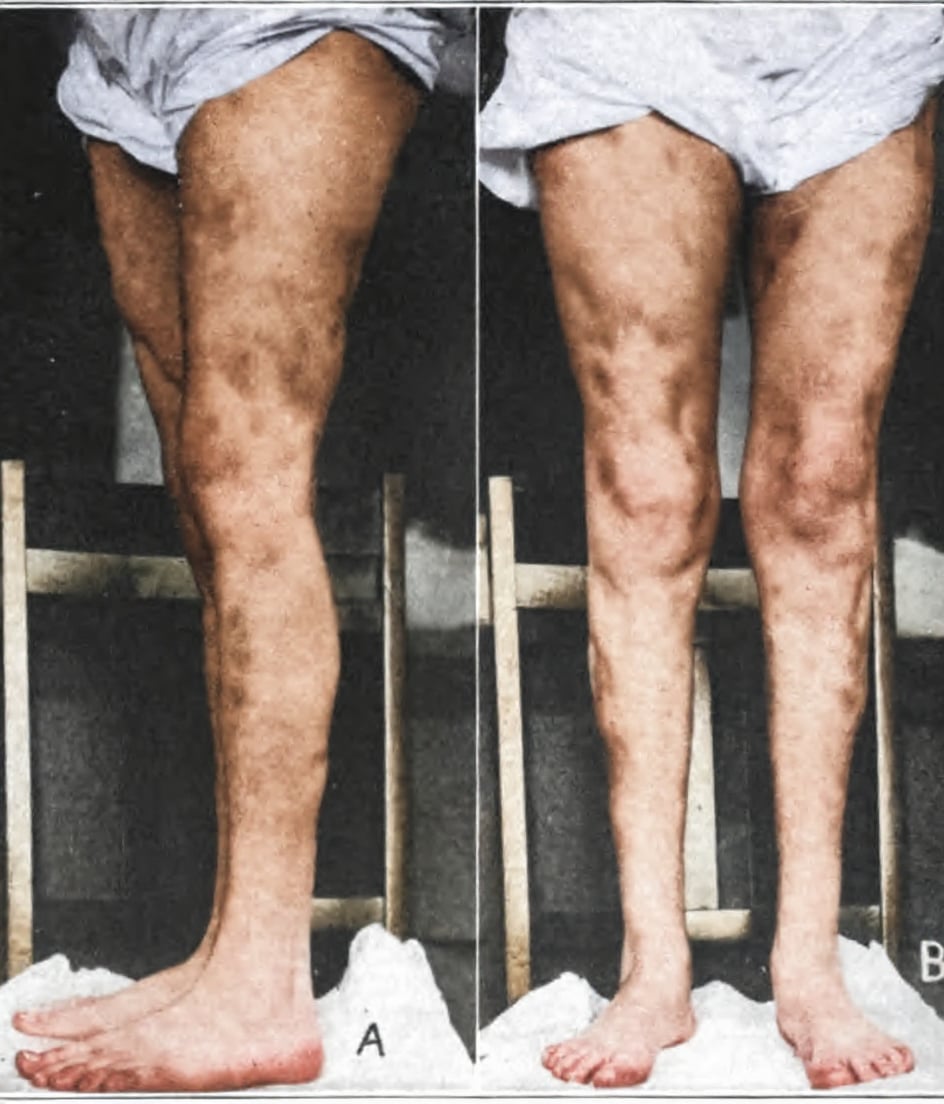
Rendu-Osler-Weber disease
Rendu-Osler-Weber disease (aka Hereditary haemorrhagic telangiectasia (HHT)) is an autosomal dominant disorder characterised by epistaxis, cutaneous telangiectasia, and visceral arteriovenous malformations (AVMs).
Characteristic lesions are telangiectases – focal dilatations of postcapillary venules and AVMs. Recurrent epistaxis often restricts daily life and AVMs can cause serious complications if located in the lungs, liver, or brain.
1907 – Weber provided a clinical description in a series of cases; and provided a summary of the world literature to date. He would later expand further with more cases in 1936
Mrs. Sarah S…has a number of bright red angiomata distributed over the face, ears, lips, tongue, mucous membrane of the mouth, and the conjunctival surfaces of the four eyelids. All these angiomata are small; many, including those on the tongue and inside of the mouth, are hardly as large as an ordinary brass pin’s head (punctiform angiomata)
On the mucous membrane inside both nostrils there are some telangiectases…On the fingers, and notably under the finger-nails, there are several minute (pinpoint) red angiomata, which I had not observed until Professor W. Osler, when he recently saw the patient, kindly drew my attention to them.
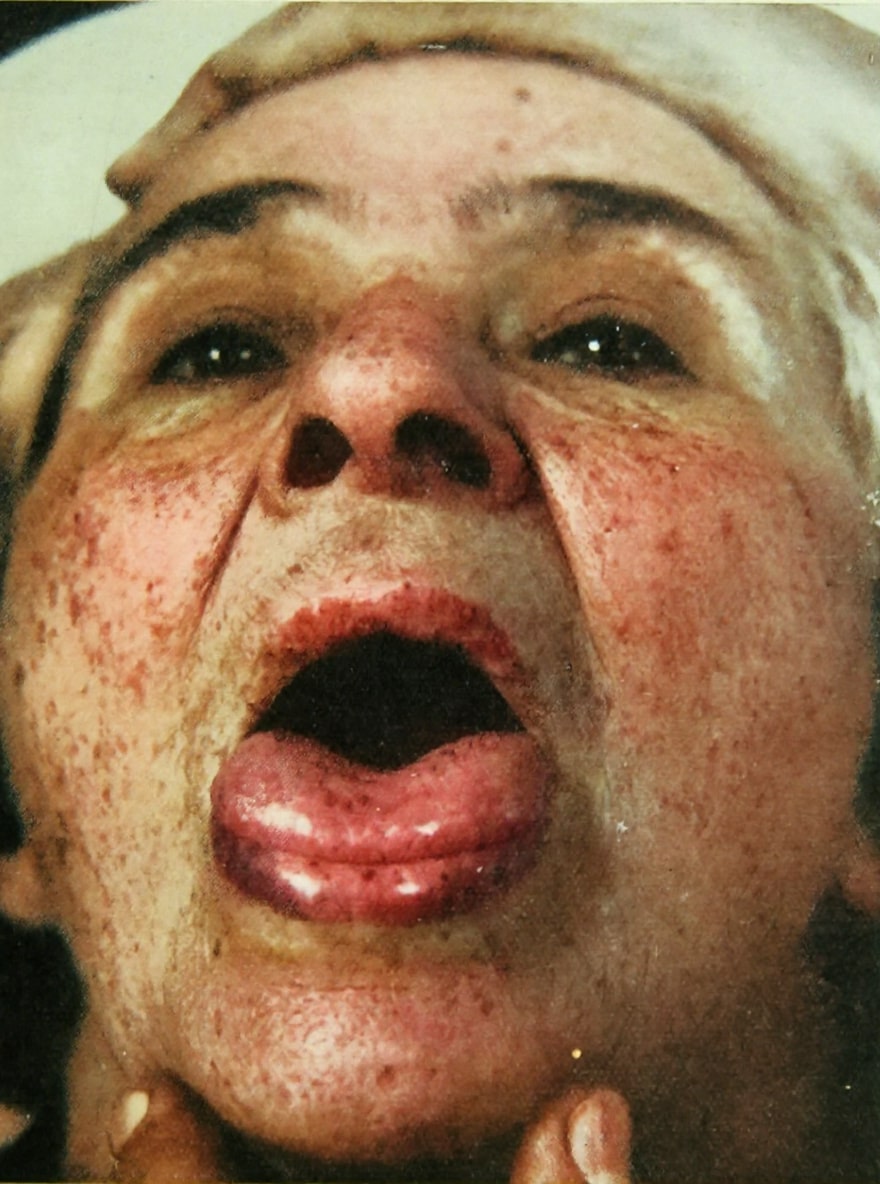
Sturge-Weber syndrome
Sturge-Weber syndrome (SWS) is a rare, congenital neurocutaneous disorder characterised by capillary-venous malformations involving the skin, leptomeninges, and ocular structures. The hallmark clinical triad includes facial port-wine stain (nevus flammeus), leptomeningeal angiomatosis, and ocular involvement, typically glaucoma.
1879 – William Allen Sturge (1850-1919) presented a 6½-year-old girl with facial port-wine stain, seizures, and glaucoma to the Clinical Society of London – A case of partial epilepsy, apparently due to a lesion of one of the vasomotor centres of the brain. He detailed focal twitching of the left side of her body at 6 months of age which later spread to the other side with loss of consciousness.
1922 – Weber demonstrated characteristic intracranial calcifications via radiographs in his article “Right-sided hemi-hypertrophy resulting from right-sided congenital spastic hemiplegia… a morbid condition of the left side of the brain, revealed by radiograms.”
1946 – Weber analysed the clinical findings and history of the disease in his book ‘Rare diseases and some debatable subjects’, in the chapter on Sturge-Kalischer disease.
The patient was indeed a remarkable sight. She was a rather obese young woman with a fleshy face, much of which, especially the left side, was scarlet or purple owing to a very large telangiectatic naevus; on the left side she had an ‘ox-eye’ (buphthalmos, congenital glaucoma), and the right half of her body was smaller than the left half and also hemiparetic, pointing to a lesion on the left side of the brain.
Because the X-ray findings by Dr James Metcalfe in my case proved the existence of a more or less calcified and apparently ‘festooned’ lesion on the surface of the left cerebral hemisphere, Krabbe
suggested the term ‘Parkes Weber-Dimitri disease’, V. Dimitri in the Argentine, having described the X-ray findings in a similar case in 1923. But in 1935 Professor Hilding Bergstrand called the condition the ‘Sturge-Weber disease, because of the priority of Dr W. Allen Sturge’s account in 1879
Weber-Cockayne syndrome
Weber-Cockayne syndrome is a form of epidermolysis bullosa simplex (EBS), an inherited genetic disorder characterised by recurrent, non-inflammatory blistering (bullous eruptions) primarily affecting the palms and soles. Blistering is typically triggered by mechanical trauma—commonly during early childhood, but cases may also present in adulthood, particularly during warm seasons. The condition affects both sexes equally.
1926 – Weber under the title, ‘Recurrent Bullous Eruption on the Feet in a Child‘, showed before the Dermatological Section of the Royal Society of Medicine a boy with the same signs and symptoms. He was 4 years of age, born to apparently normal parents, and had numerous large bullae on the soles of the feet in warm weather. Bullae had first appeared at the age of 1 year.
I suggest that the case is a mild atypical form of epidermolysis bullosa, the exciting factor being irritation of the feet in moist socks during warm weather. The points against this suggested diagnosis are:-(1) the absence of ordinary traumatism as a direct exciting agent. I failed artifically to produce a bulla by moderate rubbing of the dorsum of either foot; (2) the hands, nails and face have not yet been affected; (3) the absence of any familial history of the disease.
Major Publications
- Weber FP. A note on cutaneous telangiectases and their etiology. Comparison with the etiology of haemorrhoids and ordinary varicose veins. Edinburgh Medical Journal, 1904; 15(4): 346–349
- Weber FP. Multiple hereditary developmental angiomata (telangiectases) of the skin and mucous membranes associated with recurring haemorrhages. Lancet 1907; 170(4377): 160-162 [Rendu-Osler-Weber disease]
- Weber FP. Angioma-formation in connection with hypertrophy of limbs and hemi-hypertrophy.
British Journal of Dermatology. 1907; 19: 231-235. [Klippel-Trénaunay-Weber syndrome] - Weber FP. Hemangiectatic hypertrophy of Limbs – congenital phlebarteriectasis and so-called congenital varicose veins. British Journal of Children’s Diseases, 1918; 15: 13. [Klippel-Trénaunay-Weber syndrome]
- Weber FP. Aspects of death and correlated aspects of life in art, epigram, and poetry : contributions towards an anthology and an iconography of the subject. 1918
- Weber FP. Patches of deep pigmentation of the oral mucous membrane not connected with Addison’s disease. Quarterly Journal of Medicine. 1919; 12: 404-408. [Peutz-Jeghers syndrome]
- Weber FP. A case of relapsing non-suppurative nodular panniculitis, showing phagocytosis of subcutaneous fat-cells by macrophages. British Journal of Dermatology and Syphilis. 1925; 37: 301-311. [Pfeifer-Weber-Christian disease]
- Weber FP. Dr. Markus’s Original Case of Markus’s Syndrome (“Myotonic Pupil” with Absence of Patellar and Achilles Reflexes) shown twenty-seven and a half years ago. Proc R Soc Med. 1933 Mar;26(5):530-1. [Holmes-Adie syndrome]
- Weber FP. Haemorrhagic telangiectasia of the Osler-type «telangiectatic dysplasia» and isolated case, with discussion on multiple pulsating stellate telangiectases and other striking haemangiectatic conditions. British Journal of Dermatology. 1936; 48(4): 182-193.
- Parkes Weber F. The Sturge-Kalischer Disease. In: Rare diseases and some debatable subjects. London: Staples, 1946: 9–11.
References
Biography
- Obituary. British Medical Journal, 1962; 1(5293): 1541–1543.
- Munk’s Roll. Frederick Parkes Weber. Royal College of Physicians
Eponymous terms
Klippel-Trénaunay-Weber syndrome
- Klippel M, Trénaunay P. Du naevus variqueux ostéo-hypertrophique. Archives générales de médecine, 1900; 3: 641-672.
- Rotondo C, Corrado A, Mansueto N, Cici D, Corsi F, Pennella A, Paolo Cantatore F. Pfeifer-Weber-Christian Disease: A Case Report and Review of Literature on Visceral Involvements and Treatment Choices. Clin Med Insights Case Rep. 2020 May 27;13:1179547620917958.
Pfeifer-Weber-Christian disease (PWCD)
- Pfeifer V. Über einen Fall von herdweiser Atrophie des subkutanen Fettgewebes. Deutsches Archiv für klinische Medizin. 1892; 50: 438-449.
- Christian HA. Relapsing febrile nodular nonsuppurative panniculitis. Archives of Internal Medicine. 1928; 42: 338-351.
Rendu-Osler-Weber disease
- Sutton HG. Epistaxis as an indication of impaired nutrition, and of degeneration of the vascular system. Medical Mirror (London) 1864; 1: 769-781
- Babington BG. Hereditary epistaxis. Lancet 1865; 86(2195): 362-363
- Legg JW. A case of haemophilia complicated with multiple naevi. Lancet 1876; 108(2781): 856-858
- Rendu H. Epistaxis répétés chez un sujet porteur de petits angiomes cutanés et muqueux.
Gazette des hôpitaux civils et militaires (Lancette française) 1896; 69: 1322-1323. - Osler WB. On a family form of recurring epistaxis, associated with multiple telangiectases of the skin and mucous membranes. Bulletin of the Johns Hopkins Hospital. 1901; 12(128): 333-337.
Eponym
the person behind the name
BA MA (Oxon) MBChB (Edin) FACEM FFSEM. Emergency physician, Sir Charles Gairdner Hospital. Passion for rugby; medical history; medical education; and asynchronous learning #FOAMed evangelist. Co-founder and CTO of Life in the Fast lane | On Call: Principles and Protocol 4e| Eponyms | Books |