
![]()
Pierre Marie

Pierre Marie (1853-1940) was a French neurologist
Renowned for his work on acromegaly, aphasia, hereditary ataxias, and spinal disorders. A pupil of Jean-Martin Charcot, Marie became one of the most influential neurologists of the late 19th and early 20th centuries, mentoring a generation of clinicians who would define modern French neurology.
Pierre Marie was one of the most accomplished figures in French medicine, whose wide-ranging contributions helped shape the fields of neurology, endocrinology, and spinal pathology. A prolific researcher and rigorous clinical observer, he is remembered for his description of acromegaly in 1886, establishing it as a distinct pituitary disorder, and for his critical revisions to the classification of aphasia.
Born in Paris in 1853, Marie studied under Jean-Martin Charcot at the Salpêtrière, quickly distinguishing himself through autopsy-based research and detailed clinico-pathological correlation. He was appointed médecin des hôpitaux in 1883, and after Charcot’s death, became head of the Salpêtrière neurological service, where he trained many leading neurologists including Charles Foix, Joseph Babinski, and Georges Guillain.
Marie’s critical mind led him to challenge established doctrines, including the Wernicke–Lichtheim model of aphasia, which he reinterpreted in terms of broader cognitive dysfunctions rather than strict localisation. He was also a pioneer in describing hypertrophic pulmonary osteoarthropathy and Marie–Strümpell disease (a form of ankylosing spondylitis).
An austere but revered teacher, Marie edited the Revue Neurologique and held the Chair of Neurology at the Faculty of Medicine in Paris. Despite controversies and occasional disputes with peers, his legacy remains foundational. He died in Paris in 1940 during the German occupation.
Biographical timeline
- Born September 9, 1853 in Paris, France.
- 1870s – Studied medicine at the Salpêtrière under Jean-Martin Charcot.
- 1883 – Appointed médecin des hôpitaux.
- 1886 – Described acromegaly as a distinct endocrine disorder.
- 1892 – Became head of Charcot’s former service at Salpêtrière.
- 1897 – Founded and began editing Revue Neurologique.
- 1901 – Described hypertrophic osteoarthropathy (Marie-Bamberger syndrome).
- 1907 – Appointed Professor of Neurology at Paris Faculty of Medicine.
- 1911 – Co-described Marie-Strümpell disease (ankylosing spondylitis).
- 1910s–1920s – Mentored numerous future leaders in neurology including Foix, Guillain, and Alajouanine.
- 1930s – Continued publishing clinical studies on spinal cord disease and aphasia.
- Died April 13, 1940 in Paris.
Medical Eponyms
Marie–Foix Syndrome (1913)
Marie–Foix syndrome describes a classical brainstem stroke syndrome involving the lateral pons, presenting with ipsilateral cerebellar ataxia and contralateral hemiparesis. It reflects a crossed hemiplegic syndrome due to interruption of cerebellar and pyramidal tracts, typically attributed today to infarction in the territory of the anterior inferior cerebellar artery (AICA).
This syndrome was originally described by Pierre Marie and Charles Foix in 1913 through cases of syphilitic brainstem involvement, and further summarised in 1914. While modern terminology prefers anatomical descriptors (e.g. AICA infarct), this eponym remains of historical significance in cerebrovascular neurology.
1913 – Marie and Foix publish two case reports in La Semaine Médicale:
- One patient with pure hemicerebellar syndrome due to syphilitic mid-pontine lesion.
- Four patients with ipsilateral cerebellar and pyramidal signs. “La lésion, macroscopiquement minuscule, sectionnait tout le pédoncule cérébelleux moyen...”
1914 – Revue Neurologique publishes a detailed review summarising these findings (vol 28(14): 93-94). It classifies the syndromes into three clinical types:
- Ipsilateral cerebellar and pyramidal signs with contralateral oculomotor palsy
- Ipsilateral cerebellar and pyramidal signs with dysarthria
- Ipsilateral cerebellar and pyramidal signs with thalamic symptoms
1922 – Marie, Foix, and Alajouanine publish a separate work on cortical cerebellar atrophy, leading to long-standing eponymous confusion with Marie–Foix–Alajouanine syndrome.
Modern usage – “Marie–Foix syndrome” now generally refers to the lateral pontine stroke syndrome, often replaced in clinical settings by “AICA infarct syndrome” or “lateral pontine syndrome.”
Marie–Foix–Alajouanine syndrome (1922)
A progressive cortical cerebellar atrophy typically seen in older adults, with a strong association to chronic alcohol use. Clinically, it manifests with gait ataxia, dysarthria, intention tremor, and truncal instability — classic features of cerebellar dysfunction. Histologically, the condition involves degeneration of Purkinje cells, particularly in the anterior lobe of the cerebellum.
Originally described in 1922 by Pierre Marie, Charles Foix, and Théophile Alajouanine as l’atrophie cérébelleuse tardive à prédominance corticale, the syndrome was framed as a late-onset, predominately cortical cerebellar degeneration. While most commonly seen in alcoholics, a hereditary form was later described by Richter in 1950, suggesting an autosomal dominant inheritance in select families.
In modern neurology, the term is used less frequently, with most cases now subsumed under alcoholic cerebellar degeneration or spinocerebellar ataxias (SCAs), depending on aetiology and family history.
Charcot-Marie-Tooth disease (1886)
Charcot–Marie–Tooth disease (CMT) is one of the most common inherited neurological disorders, encompassing a group of genetically and clinically heterogeneous peripheral neuropathies.
1886 – Jean-Martin Charcot (1825-1893) and Pierre Marie describe five patients with a familial, progressive atrophy of distal lower limbs – Sur une forme particulière d’atrophie musculaire progressives , proposing a spinal origin.
Though often criticised for attributing the disorder to myelopathy rather than neuropathy, they acknowledged diagnostic uncertainty and refrained from definitive localisation — a cautious insight given the limited tools of the time.
Original
English
Est-ce done a une myelopathie que nous avons affaire? est-ce a une nevrite multiple peripherique? Ici, il faut bien l’avouer, la question devient beaucoup plus delicate, en presence surtout de ces cas ou il a existe soit des douleurs, soit des troubles divers de la sensibilite; bien que jusqu’a un certain point l’hypothese d’une myelopathie nous paraisse preferable il nous
semble difficile de se prononcer d’une facpn absolue
Is this a myelopathy? or multiple peripheral neuritis? Here, we must say, the question is more complicated, especially in the presence of cases with either pain or varied sensory disorders; although, up to a certain point, we prefer the hypothesis of myelopathy, it seems difficult to come to a firm conclusion.
Bamberger–Marie syndrome (1889)
Also known as Piere Marie-Bamberger syndrome or hypertrophic osteoarthropathy (HOA), is a rare clinical entity characterized by digital clubbing, periostosis of long bones, and synovial effusions. It exists in two forms: primary HOA, a hereditary and less common form (also termed pachydermoperiostosis), and secondary HOA, which arises in association with systemic conditions — most notably intrathoracic malignancies such as non-small cell lung carcinoma.
The condition is primarily paraneoplastic in adults, where its presence may precede the detection of a pulmonary neoplasm. HOA affects both genders but is more common in adult males, particularly those with underlying lung disease.
1889 – Eugen von Bamberger (1858–1921) presents to the Vienna Medical Society his observations of digital clubbing in non-tuberculous bronchiectasis, published in Wiener klinische Wochenschrift
1890 Pierre Marie publishes a seminal 36-page monograph describing what he terms l’ostéo-arthropathie hypertrophiante pneumique, citing Bamberger’s observations as a precedent.
Original
English
…c’est un fait d’observation vulgaire que dans la tuberculose les doigts subissent fréquemment la déformation en baguettes de tambour. De même tout récemment (Société des Médecins de Vienne, séance du 8 mars 1889), le professeur Bamberger disait avoir observé dans plusieurs cas les doigts hippocratiques chez des individus atteints uniquement de dilatation des bronches sans tuberculose. Or ce que je soutiens c’est que la déformation qui fait l’objet du présent travail est, soit au point de vue morphologique, soit au point de vue pathogénique, un phénomène du même ordre que les doigts hippocratiques
…it is a common observation that in tuberculosis, the fingers frequently undergo the drumstick deformation. Similarly, very recently (Vienna Medical Society, meeting of March 8, 1889), Professor Bamberger reported having observed Hippocratic fingers in several cases in individuals suffering only from bronchial dilation without tuberculosis. The deformity is from a morphological or pathogenic point of view, a phenomenon of the same order as hippocratic fingers.
Marie cites Nikolaus Friedreich (1825-1882) and his 1868 paper – Hyperostose des gesamten Skelettes) in brothers Karl and Wilhelm Hagner in the context of bone and joint pathology described as potentially acromegalic.
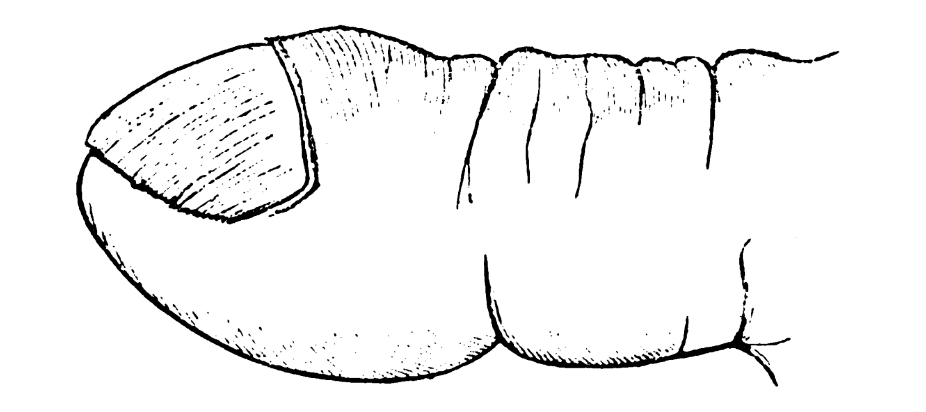
However, Marie differentiates the periosteal changes seen in hypertrophic osteoarthropathy from those of acromegaly, highlighting:
- In HOA, periostosis occurs primarily at the diaphyses and metaphyses of long bones.
- Joint effusions are common, but soft-tissue overgrowth and jaw enlargement—hallmarks of acromegaly—are absent.
- The HOA processes are symmetrical and closely tied to pulmonary disease, rather than the endocrine-driven bone expansion seen in acromegaly.
This sharp clinical distinction demonstrates Marie’s careful morphological and pathogenetic classification, years before modern endocrinology could confirm hormonal causes.
Marie-Strümpell disease (1884)
Marie-Strümpell disease, also known as ankylosing spondylitis (AS), is a chronic, progressive inflammatory spondyloarthropathy predominantly affecting the axial skeleton—particularly the spine and sacroiliac joints. Characterized by back pain, stiffness, and eventual vertebral fusion, it often manifests in young adult males (age 15–35) and is closely associated with HLA-B27 positivity.
Clinically, the disease presents with inflammatory back pain, reduced spinal mobility, and can involve enthesitis, uveitis, and cardiovascular complications. Radiographic findings and modified New York Criteria are key to diagnosis.
1884 – Adolf von Strümpell (1853–1925) briefly described the disease in his chapter on Der chronische Gelenkrheumatismus (chronische Polyarthritis) und die Arthritis deformans, where he observed progressive stiffening and fusion of the spinal joints.
Original
English
Als ein merkwürdiges und, wie uns scheint, eigenartiges Leiden mag hier beiläufig noch diejenige Erkrankungsform erwähnt werden, bei welcher es ganz allmählich und ohne Schmerzen zu einer vollständigen Ankylose der ganzen Wirbelsäule und der Hüftgelenke kommt, so dass Kopf, Rumpf and Oberschenkel fest mit einander verbunden und vollkommen steif sind, während alle übrigen Gelenke ibre normale Bewegliehkeit behalten. Dass hierdurch ganz eigenthümliche Modificationen der Körperhaltung und des Ganges entstehen müssen, liegt auf der Hand. Wir selbst haben zwei ganz gleiehartige Fälle dieser eigenthümlichen Krankheit gesehen.
As a remarkable and, as it seems to us, peculiar ailment…in which there is a complete ankylosis of the whole spine and the hip joints very gradually and without pain, so that the head, trunk and thighs are firmly together connected and perfectly rigid while all remaining joints retain their normal mobility. It is obvious that this results in very peculiar modifications of posture and gait. We ourselves have seen two very similar cases of this peculiar disease.
1893 – Vladimir M. Bekhterev (1857–1927) published Steifigkeit der Wirbelsäule und ihre Verkrümmung als besondere Erkrankungsform, detailing spinal rigidity and deformity.
1897 – Strümpell published Bemerkung über die chronische ankylosirende Entzündung der Wirbelsäule und der Hüftgelenke detailing another case study of a patient with the disease in more detail., reinforcing chronic axial arthropathy.
1898 – Marie made several foundational distinctions that shaped the clinical identity of ankylosing spondylitis in his publication on spondylose rhizomélique. He noted rigidity of the chest, the spine being ‘fait rigide comme un baton‘; and autopsy demonstratged the spine had ‘L’ossification tout particulièrement localisée aux ligaments; aux bourrelets et aux menisques articulaires‘.
Marie essentially defined a distinct clinical entity and isolated the disease from other spinal disorders emphasising an ascending pattern of spine involvement moving from the sacroiliac region upward to the cervical spine. He differentiated it from osteoarthritis and rheumatoid arthritis, noting:
- Symmetrical fusion of vertebral bodies and sacroiliac joints
- Sparing of large peripheral joints in early disease
- Absence of erosive changes typically seen in rheumatoid arthritis
Marie also correlated clinical and radiologic features (even before widespread use of X-rays), describing:
- Persistent, inflammatory low back pain and stiffness
- Marked limitation of spinal mobility
- Kyphotic deformity in advanced stages
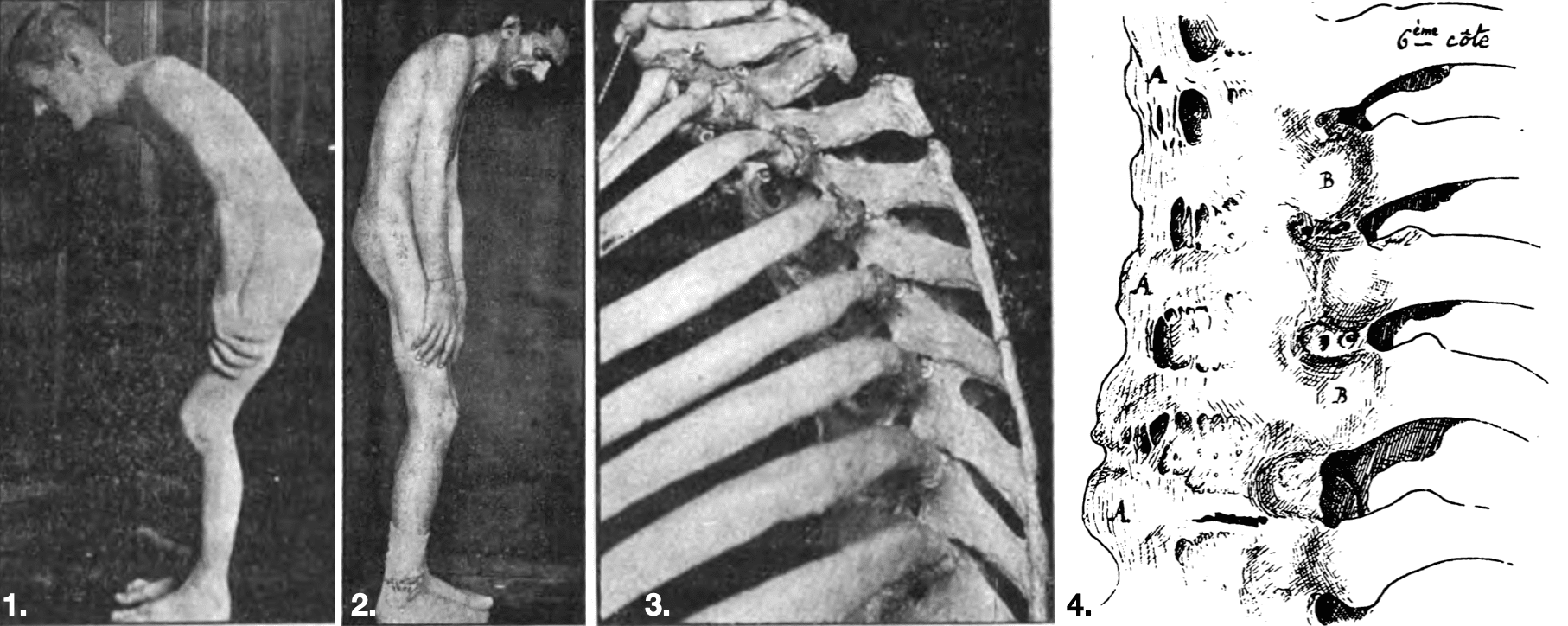
3. Ossification of the supraspinous ligament
4. A: Ossification of the large anterior ligament; B: Heads of the vertebrae connected to the vertebrae by ossification of their ligaments.
1920s–1940s: Recognition of sacroiliac involvement by Forestier, Scott, and Hare. Radiologic correlation with disease onset established.
Acromegaly and Endocrinology (1886)
Pierre Marie was the first to coin the term “acromegaly” in 1886, formally distinguishing it as a unique clinical entity. In his original description, he defined the condition as characterized primarily by hypertrophy of the hands, feet, and face, proposing the name “acromegaly” (from Greek “akron” = extremity and “megas” = large) to reflect this hallmark feature.
Marie emphasized that while the hypertrophy was initially most visible in the extremities, it represented a broader systemic disease. He differentiated acromegaly from other conditions such as myxedema, Paget’s disease, and leontiasis ossea, making a lasting contribution to clinical endocrinology.
Original
English
1° Il existe une affection caractérisée surtout par une hypertrophie des pieds, des mains et du visage, que nous proposons d’appeler acromégalie, c’est-à-dire hypertrophie des extrémités (non pas qu’en réalité, les extrémités soient seules atteintes pendant toute la durée de la maladie, mais parce que leur augmentation de volume est un phénomène initial et constitue le trait le plus caractéristique de cette affection).
2° L’acromégalie est tout à fait distincte du myxcedème et de la maladie de Paget (ostéite déformante), ainsi que de la leontiasis ossea de Virchow.
1° There is a condition characterized mainly by hypertrophy of the feet, hands and face, which we propose to call acromegaly, that is to say hypertrophy of the extremities (because the increase in volume of teh extremities is an early sign and constitutes the most characteristic feature of this condition).
2° Acromegaly is quite distinct from myxoedema and Paget’s disease (osteitis deformans), as well as from Virchow’s leontiasis ossea.
While earlier physicians (e.g., Johannes Wier in 1567, Alibert in 1822) had described similar phenotypes under various names, Marie’s work unified the clinical picture under one term. In the years that followed, Marie and his colleagues (notably J.D. Souza-Leite and G. Marinesco) further elucidated the pathological correlation with pituitary enlargement.
By the end of the 19th century, it was increasingly recognized that a pituitary adenoma causing hyperfunction of the gland was the underlying cause of acromegaly. This insight laid the groundwork for the later understanding of the growth hormone (GH)–IGF-1 axis, marking Marie’s work as foundational in the evolution of neuroendocrinology.
Major Publications
- Marie P. Contribution à l’étude et au diagnostic des formes frustes de la maladie de Basedow. Thèse de doctorat, Paris, 1883
- Charcot JM, Marie P. Sur une forme particulière d’atrophie musculaire progressive souvent familiale débutant par les pieds et les jambes et atteignant plus tard les mains. La Revue de médecine 1886; 6: 96-138 [English translation: Concerning a special form of progressive muscular atrophy: often familial starting in the feet and legs and later reaching the hands. Arch Neurol. 1967; 17(5):553-557] [Charcot-Marie-Tooth disease]
- Marie P. Sur deux cas d’acromégalie; hypertrophie singulière non congénitale des extrémités supérieures, inférieures et céphalique. La Revue de médecine 1886; 6: 297–333. [acromegaly]
- Brissaud E, Marie P. De la déviation faciale dans l’hémiplégie hystérique. Le Progrès médical, 1887; 5: 84-85. [Briquet’s syndrome I]
- Marie P. De l’ostéo-arthropathie hypertrophiante pneumique. Revue de médecine 1890; 10: 1-36 [Bamberger–Marie syndrome]
- Marie P. Leçons sur les maladies de la moelle. 1892 [Lectures on diseases of the spinal cord, 1895]
- Marie P. Sur l’hérédo-ataxie cérébelleuse. Clinique des maladies nerveuses. La semaine médicale, 1893; 13: 444-447. [Marie’s ataxia]
- Marie P. Sur la contraction réflexe des adducteurs de la cuisse déterminée par la percussion du tendon rotulien du côté opposé. Bulletins et mémoires de la Société médicale des hôpitaux de Paris, 1894; 11: 246-248. [Marie’s crossed adduction reflex]
- Marie P. Sur la spondylose rhizomélique. Revue de médecine, 1898, 18: 285-315. [Marie-Strümpell disease]
- Marie P. Présentation de malades atteints d’Anarthrie par lésion de l’Hémisphére gauche du Cerveau. Bulletins et mémoires de la Société médicale des hôpitaux de Paris, 1907; 1: 864-865
- Marie P, Foix C. Sur la retrait réflexe du membre inférieure provoqué par la flexion forcée des orteils. Revue neurologique, 1910; 26(14): 121-123. [Marie-Foix manoeuvre and reflex]
- Marie P, Foix C. Les réflexes d’automatisme medullaire. Revue neurologique, 1912; 23(10): 657-676 [Marie-Foix manoeuvre and reflex]
- Marie P, Foix C. Hémisyndrome cérébelleux d’origine syphilitique: hémiplégie cérébelleuse syphilitique. La Semaine Médicale. 1913; 33(2): 13–15. [Marie-Foix syndrome]
- Marie P, Foix C. Atrophie isolée de l’éminence thénar d’origine névritique. Rôle du ligament annulaire dans la localisation de la lésion. Revue Neurologique 1913; 26: 647-649
- Marie P, Foix C, Alajouanine T. De l’atrophie cérébelleuse tardive à prédominance corticale.
Revue neurologique 1922; 38(7): 849-885 and 1922; 38(8): 1082-1111. [Marie-Foix-Alajouanine syndrome] and [Marie-Foix syndrome] - Foix C, Chavany JA, Marie J. Diplégie facio-linguo-masticatrice d’origine sous-corticale sans paralysie des membres (contribution à l’étude de la localisation des centres de la face du membre supérieur). Revue Neurologique. 1926; 1: 214–219. [Foix–Chavany–Marie syndrome]
References
Biography
- Cohen H. Pierre Marie 1853-1940. Proc R Soc Med. 1953 Dec;46(12):1047-54.
- Poirier J, Chretien F. Pierre Marie (1853-1940). J Neurol. 2000 Dec;247(12):983-4.
- Pearce JM. A note on Pierre Marie (1853-1940). J Neurol Neurosurg Psychiatry. 2004 Nov;75(11):1583.
- Loriaux DL. Pierre Marie (1853–1940). The Endocrinologist, 207; 17(5): 243.
- Poirier P, Poirier J. Pierre Marie (1853-1940) : dans l’intimité d’un neurologue remarquable. Mollat 2023
- Fresquet JL. Pierre Marie (1853-1940). Historia de la Medicina
- Portrait. Pierre Marie. Wellcome collection
Eponymous terms
- Borak J. Significance of the Sacroiliac Findings in Marie-Strümpell’s Spondylitis. Radiology 1946; 47(2): 128–141.
- Düster P. Pierre-Marie-Bamberger Syndrom – Ein paraneoplastisches Syndrom beim Bronchialkarzinom – Ein Fallbericht – [Pierre-Marie-Bamberger syndrome – a paraneoplastic syndrome of lung cancer – a case report]. Zentralbl Chir. 2002 Jan;127(1):59-61
- Koliakos E, Chappalley D, Kalogiannis E, Sgardello S, Christodoulou M. Pierre-Marie Bamberger Syndrome Leading to the Diagnosis and Surgical Treatment of a Localized Lung Cancer. Cureus. 2023 Nov 18;15(11):e48991.
Eponym
the person behind the name
BA MA (Oxon) MBChB (Edin) FACEM FFSEM. Emergency physician, Sir Charles Gairdner Hospital. Passion for rugby; medical history; medical education; and asynchronous learning #FOAMed evangelist. Co-founder and CTO of Life in the Fast lane | On Call: Principles and Protocol 4e| Eponyms | Books |