
![]()
A Really Tricky ABG?
aka Metabolic Muddle 013
A 30 year-old female was admitted to the ICU following a traumatic brain injury. Her ABG was normal on arrival. Since admission her condition has deteriorated and she now has circulatory failure requiring 100mcg/min of noradrenaline IV.
This is her arterial blood gas now:
Questions
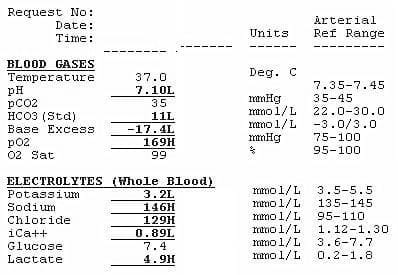
Q1. Describe the ABG
Description
- Metabolic acidemia (pH 7.1 with HCO3 11)
- Incomplete respiratory compensation
— Expect PaCO2 = 25 mmHg based on Winters’ formula: (HCO3 x 1.5) +8 (±2) - Low-Normal anion gap
— AG = Na – (HCO3 + Cl) = 146 – (11 + 129) = 6 mmol/L
— normal AG depends on the reference range provided by the lab that performed the blood test - Hypernatremia and hyperchloremia
- Hypocalcemia and hypokalemia
- Hyperlactemia
Albert MS, Dell RB, Winters RW. Quantitative displacement of acid-base equilibrium in metabolic acidosis. Annals of Internal Medicine. 1967; 66 (2): 312–22.
Q2. Interpret this ABG
Interpretation
- Triple acid-base disturbance – NAGMA + HAGMA (i.e. hyperlactemia, even though the anion gap is calculated as normal) + respiratory acidosis
- The possible causes of NAGMA, using an ABCD approach are:
— Addison’s (causes hypotension, but Na should be low and K should be high)
— Bicarbonate loss from the GI tract (e.g. diarrhoea, fistula) or the kidneys (e.g. renal tubular acidosis)
— Chloride excess (e.g. hypertonic saline administration in TBI)
— Drugs (e.g. acetazolamide) - Hypokalemia, hyperchloremia in the presence of a marked NAGMA is suggestive of type 1 or distal renal tubular acidosis. Potassium would be even lower if there wasn’t a metabolic acidosis (K moves out of cells in exchange for H+)
- Hyperlactemia is likely due to circulatory failure other possibilities are seizures and the beta-2 agonist effects of catecholamines (unlikely with noradrenaline unless very high doses used, leading to loss of receptor selectivity)
- Hypocalcemia — the specific cause is uncertain (see below), but its presence is important as correction may (and in fact did) improve the patient’s circulatory failure.
- Respiratory acidosis – the patient is sedated and mechanically ventilated after TBI, so the patient is unable to compensate for metabolic acidosis by hyperventilation in the usual manner. Low-normal PCO2 is favoured in TBI to prevent cerebral vasoconstriction, which occurs at lower PCO2, from exacerbating brain injury.
Commentary from Paul Young
Further comment
Many people suggested normal saline resuscitation as the cause, this is not possible (certainly not in isolation). No-one develops a bicarbonate of 11 after resuscitation with normal saline no matter how much you give them. The lactic acidosis is mild and AG is not high so the results suggest that there is a secondary NAGMA (i.e not just NaCl). On the basis of the severe shock Addison’s would be a good suggestion but RTA should be very high up the differential. GI loss (if this was the cause) would generally be obvious. Diabetes insipidis is clinically likely (given **** load of Noradrenaline and severe TBI). Propofol infusion syndrome is exceptionally rare but this is the setting in which it is typically seen.
So, we cannot exclude these possible contributors to circulatory failure:
Diabetes inspidus
- primarily a clinical diagnosis (dehydration, sustained dilute polyuria) confirmed by lab testing (e.g urine specific gravity of 1.005 or less and a urine osmolality less than 200 mOsm/kg, plasma osmolality > 287 mOsm/kg).
- it fits the clinical scenario of severe TBI, and even without urea the calculated plasma osmolality is nearly 300.
Propofol infusion syndrome
- can cause of lactic acidosis and circulatory failure.
- TBI patients can get repeated boluses in addition to the propofol infusion used to maintain sedation keep ICPs low, so they can be at risk
- can also cause rhabdomyolysis which can contribute to hypocalcemia
Of course the patient could have other causes of circulatory failure such as hemorrhage from associated injuries.
An interesting feature of this blood gas is that the anion gap is not elevated despite the presence of hyperlactemia. We should note that calculating anion gap is only about 50% sensitive for detecting the presence of a HAGMA! Reasons for this can include:
- hypoalbuminemia is most common
- also consider coexistent causes of a low anion gap
- Decrease in unmeasured anions (albumin, dilution)
- Increase in unmeasured cations (multi-myeloma, hypercalcaemia, hypermagnesaemia, lithium OD, bromide OD, polymixin B)
- Non random analytical errors (increased Na+, increased viscosity, iodide ingestion, increased lipids)
Q4. What is the Strong Ion Difference?
Answer and interpretation
Using the physicochemical/ Stewart approach, the Strong Ion Difference (normal is 38) is markedly low (Na – Cl = 146 – 129 = 17). The causes of this are the same as the causes of NAGMA (see also EMCrit — Acid Base in the Critically Ill – Part I).
We don’t have the albumin so we can’t calculate a corrected anion gap or strong ion gap, but we already know the lactate is high and that the NAGMA is marked. Delta gap or ratio is best calculated using a corrected anion gap and can be performed in the setting of a metabolic acidosis to check for a mixed disorder – but we already know we have a triple acid-base disorder based on the initial analysis.
Q5. What about the low calcium?
Answer and interpretation
The hypocalcemia was important in this case, because calcium replacement markedly decreased noradrenaline requirements and improved circulatory failure. The precise causes are unclear, and are often multifactorial in ICU patients. Here are some of the more common possibilities in ICU:
- Hypomagnesemia
- Hyperphosphatemia e.g. rhabdomyolysis
- Vitamin D deficiency or resistance
- diuretics e.g. frusemide (also low K and low Mg)
- renal failure
Other less likely possibilities (if you feel like dementing yourself further):
- overhydration – e.g. fluid resuscitation to maintain MAP and CPP
- citrate toxicity from PRBCs – rare, unless liver or kidney disease impairs citrate clearance
- PTH deficiency or resistance (an be due to low Mg)
- Multifactorial enhanced protein binding and anion chelation
- pancreatitis
- hungry bone syndrome
- longterm anticonvulsants (e.g. phenytoin-mediated CYP450 induction increases Vit D catabolsim)
- radiographic contrast dyes (e.g. EDTA chelates, gadolinium can falsely lower calcium shortly after imaging)
Hypoalbuminemia can cause hypocalcemia but it doesn’t affect ionized calcium, so can’t be the cause in this case.
So what was the underlying metabolic disturbance?
Further Reading
In this case the patient did not have excessive GI fluid losses, was not on any drugs that would contribute to NAGMA and had a recent short synACTHen test excluding adrenal insufficiency. An acid load test confirmed the presence of incomplete type 1 or distal renal tubular acidosis!?! (hence the title Really Tricky ABG – the biggest clue of all!).
Features of dRTA include:
- urine pH >5.5 (after acid load test)
- increased urine anion gap ([Na+]+ [K+] – [Cl-]) consistent with decreased urine NH4+
- hypercalciuria (to my knowledge this doesn’t tend to cause hypocalcemia however)
Further discussion, points of clarification, etc etc, are — as always — most welcome!
CLINICAL CASES
Metabolic Muddle
Chris is an Intensivist and ECMO specialist at The Alfred ICU, where he is Deputy Director (Education). He is a Clinical Adjunct Associate Professor at Monash University, the Lead for the Clinician Educator Incubator programme, and a CICM First Part Examiner.
He is an internationally recognised Clinician Educator with a passion for helping clinicians learn and for improving the clinical performance of individuals and collectives. He was one of the founders of the FOAM movement (Free Open-Access Medical education) has been recognised for his contributions to education with awards from ANZICS, ANZAHPE, and ACEM.
His one great achievement is being the father of three amazing children.
On Bluesky, he is @precordialthump.bsky.social and on the site that Elon has screwed up, he is @precordialthump.
| INTENSIVE | RAGE | Resuscitology | SMACC
should one of dRTA features be hypercalciuria rather than hypocalciuria? Thanks for the article!
Thanks Shawn – you are correct – we have corrected the error.
Cheers, Chris