
![]()
Malaria vaccine
Introduction

The malaria disease is caused by a Plasmodium protozoal parasite, post proboscis penetration by an infected Anopheles mosquito. Around 3 billion people reside in endemic areas leading to an estimated 250 million cases and 0.9 million fatalities annually (1-4). Though confined to sub-tropical regions, its vector-borne and sometimes zoonotic characteristics make it a difficult disease to eradicate from endemic regions. In areas of endemicity, those who succumb to the disease are often pregnant women and children under the age of five (5). Further, these areas are impoverished and logistically remote, making adequate treatment for this locally relatively common illness difficult. Whilst some medicines active against the Plasmodium have been developed, the parasite has acquired resistance to many (6). The effective control of malaria transmission reduces the likelihood of the parasite’s further developing resistance to existing therapies, from a public health point of view, this would be the optimum solution to countering the disease threat. Given that the locations where malaria is most prevalent are remote, stability of a vaccine and the logistics of supplying it will be key considerations alongside the efficacy of any such preventative therapy.

Malaria presents as a spectrum of clinical disorders, varying from person to person, and dependent on: region, parasite causing the infection and how compliant an infected patient is with chemotherapy (1). For the purposes of this assignment, the parasite of interest shall be Plasmodium falciparum which produces the most severe malarial disease condition, cerebral malaria. The main foci of the P. falciparum are found in sub-Saharan Africa (7). Though covering a large geographical area, the parasites exhibit a regional clustering of their antigens which accounts for the loss of protective immunity in individuals who move between regions (6). This places further restrictions on vaccine design. To optimise the economic case for any vaccine, the formulation must have sufficient geographical coverage to make investment in its development viable.
The Plasmodium falciparum parasite throughout its lifecycle actively evades or utilises host immune responses to its benefit. Low parasite numbers initially act to reduce its visibility to the immune system (8). The intracellular, hepatocyte life stage only allows slow feeding of parasite antigen to the cytotoxic cells which are essential in clearing infection (4). Upon emergence from the hepatocyte, thousands of merozoites are enveloped in host cell membrane components making them indistinguishable from host cells to the immune system, finally bursting out from the protective host provided cloak in the lung, to infect erythrocytes (4). Erythrocytes aren’t nucleated and lack major histocompatability complex (MHC) molecules and are therefore unable to present parasite antigens to the CD8+ T-cells, a branch of the adaptive immunity specialised in destroying infected cells, making them important a in establishing sterilising immunity. Immune responses lead to the upregulation of intracellular and vascular cell adhesion molecules (ICAM-1 and VCAM-1 respectively) which allow for the migration of leukocytes to the site of antigen detection. Blood stage P. falciparum is expresses the P. falciparum erythrocyte membrane protein 1 (PfEMP-1) gene which binds to ICAM-1 and VCAM-1 for cytoadherence (4). Cytoadherence causes much of the serious pathology observed in P. falciparum by occluding blood vessels, the cause of cerebral malaria. Additionally, this sticking to the lumen of the blood vessels prevents splenic macrophages (which monitor for blood borne pathogens)from phagocytosing the infected erythrocytes, further delaying immune responses to the parasite. Due to the difficulty in “seeing” P. falciparum throughout its lifecycle and the serious consequences associated with infection, a vaccine is a much desired public health intervention in breaking transmission and protecting the host from serious sequelae.
Pre-Vaccine Achievements and the First Vaccine
The development of a malaria vaccine has required scientists to study the malaria parasite, its life-cycle and the interactions between host and parasite that lead to immunity. It wasn’t until the 1963 that the complete rodent malaria life-cycle was elucidated (8), with this breakthrough came the ability to create a disease model (4). Rodents being mammals have similar lymphatic systems and immune systems which allowed for better immunological characterisation of pathogenesis and immunology. Additionally, it allowed for the testing of experimental anti- malaria vaccine ideas by 1967 (8).
Malaria’s life cycle can be divided into a pre-erythrocytic stage, an erythrocytic stage and a sexual stage. At each stage different immunological responses play protective, sterilising, and “altruistic” effects (2, 4, 9, 10). Each form of immunity plays a role in the overall strategy to control malaria as a public health issue, but play a different role from a clinical perspective and are mediated by different effector mechanisms.

Sterilising immunity can be defined as complete clearance of a pathogen from the host organism. Benefits of sterilising immunity have both public and individual health significance as the Plasmodium pathogen cannot go on to cause further disease, nor can it mature to the sexual and transmissible stage of its life-cycle (4, 6, 11). This may seem like the silver bullet, or pinnacle of research and development, but it was the original concept of X-irradiated sporozoites (RAS) in 1970 that first achieved this goal and was the gold-standard for malaria vaccinology for many decades, achieving 94% protection for 10 months (6, 8). Ideally a vaccine mimics the normal infection process to provide artificial immunity that will protect against a similar albeit natural challenge. Studies of the immunity generated by the RAS vaccine pointed to CD8+ T-cells acting as the main form of cytotoxic effector cell (4, 6, 8, 9). Additionally, the liver is an independent site for the priming of CD8+ T-cells, individuals repeatedly infected with malaria have detectable CD8+ T-cells that recognise the sporozoite form’s major protein circumsporozoite protein (CSP), which given Plasmodium’s association with the host endoplasmic reticulum, makes for good peptide presentation and elimination of infected hepatocytes (4). Killing of the infected hepatocytes presenting CSP antigen on their major histocompatability complex I (MHC I) it is hypothesised, is mediated by T-cells and NK cells producing interferon gamma (IFN-γ), which in turn stimulates the expression of inducible nitrous oxide synthase (iNOS) (4). Although iNOS alone is dispensable in the killing of hepatocytes, it is essential in achieving sterilising immunity, along with a complex combination of CD4+, CD8+ and antibodies are involved in protecting individuals at this stage (8).
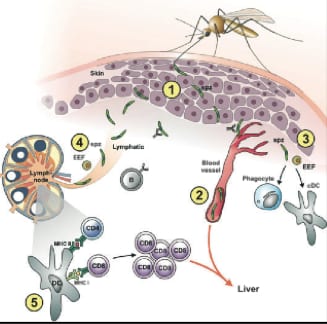
Effective malaria immunology comes down to a game of numbers. Whilst the X-irradiated sporozoites were able to induce immunity, the numbers required (in the thousands) dwarfs the 10-100 sporozoites deposited by a highly infected mosquito (4). Further, difficulties in culturing sporozoites to produce such a vaccine make the attenuated sporozoite proposition one that isn’t viable for the production of a mass-produced population level intervention (8). Indeed, the low numbers of sporozoites injected during a mosquito bite will mean a reduced encounter rate with antigen presenting cells (APCs) and therefore a slow build up of an immune response to the sporozoite phase over time with new infections, as is implied by immunity gained through the infection/treatment method (6). Again, this proposed method requires chloroquine and other anti-malarials to control the infection. Given the number of deaths occurring each year from malaria as a result of: parasite resistance, insufficient immunity and the inability to produce enough sporozoites, this method still leaves much to be desired.
Figure 1: 1. Injection of sporozoites via Anopheles mosquito into the vertebrate host. The sporozoites migrate to blood capillaries 2. Some sporozoites do not reach the liver and incompletely mature in the skin. 3. Innate immune cells (phagocytes) will clear some parasites remaining in the dermis. 4. Sporozoites drained by the lymphatic system will arrive at macrophage dense lymph nodes. 5. Macrophages and dendritic cells will present parasite epitopes and trigger the protective T-cell responses. Adapted from Hafalla.
Subunit Vaccines
With the advent of modern molecular biology in the 1980s, came the ability to express proteins which answered the “where to from here?” question being asked by many malaria immunologists. The expression of malarial antigens, the first of which was CSP (8). As mentioned, CSP vaccines act at a sufficiently early stage to generate sterilising immunity, however, due to the low doses naturally encountered, immune recognition of this antigen is slowly acquired. Twenty years after the 1973 protective malaria vaccine, the Spf66 fusion protein of the CSP and a blood stage malaria antigen was trialled as a vaccine, the approach failed (9). However, this did not mark the end of the fusion protein approach, the current and most promising progeny of the CSP sub-unit vaccine is the currently clinically evaluated RTS,S family of vaccines (5, 8, 12, 13). The RTS,S is a fusion protein of the CSP and a hepatitis B surface antigen protein (HBsAg) expressed in fungi (5, 8, 12, 13), HBsAg is a highly immunogenic component used for this purpose (12). The RTS,S/A01E formulation reached
59.1% vaccine efficacy over 17 months of phase III trials after all three doses were administered (5). Studies revealed that the the vaccine did produce a humoural response and that the geometric mean titre of anti-CSP antibodies did correlate with disease protection, further the cellular immunological process also plays a large part in this vaccine’s anti-malarial immunity. The CSP protein is responsible for parasite binding to hepatocytes’ highly sulphated heparan sulphate proteoglycans (HSPGs), blocking the CSP protein through high avidity antibodies prevents parasite binding to HSPGs and therefore prevents through an as yet uncharacterised mechanism the parasites’ gaining entry into hepatocytes (4). Ironically, this binding interaction responsible for finding hepatocytes may suppress liver macrophages, Kupffer cells, from performing their phagocytic function and further impede the immune system’s developing adaptive immunity to CSP. This may explain the attenuation of disease observed in the RTS,S vaccine trials. Studies are still examining the role of cellular immunity in the RTS,S vaccine.
Other teams’ approaches to malarial vaccination have also been produced promising fusion proteins. Blood stage fusion vaccines like GMZ2 which were being trialled in 2010, fuse the glutamate rich protein (GLURP27-500) and merozoite surface protein (MSP3212-380) to mimic pathogen induced semi-immunity (10). MSP based approaches previously failed due to regional polymorphisms in this surface exposed antigen (4, 6). The peptide epitopes selected in the manufacture of this vaccine attempt however were chosen due to the high degree of conservation exhibited in these regions of the respective proteins (10). Both GLURP and MSP antibodies are found in high levels in children protected from clinical malaria, further the antibodies generated are mainly cytophilic IgG1 and IgG3 (14). Cytophilic antibodies bind to the Fcγ receptors on circulating monocytes, which may further present other blood stage antigens to adaptive immune effector cells, creating a more diverse range of protective antibodies. Whilst the vaccine did not produce sterilising immunity, IgG3 was a strong predictor of protection from clinical malaria (14).
Recombinant Viral Vaccines
Recombinant viral vectors have been investigated for protective immunity against clinical malaria (2, 3). In 2008, successful attempts were made to fuse adenovirus and vaccinia virus with MSP-142 (AdM42). The adenovirus fusion construct was used to prime immunity, and the vaccinia to boost it. AdM42 when tested in mice protected 100% of those challenged with 104 sporozoites (2). Protection elicited by this vaccine was humoural and also CD8+ T-cell mediated rendering the mice not only protected against but also blood and liver stages of the infection. The multiple mechanisms of resistance were able to clear the Plasmodium parasites altogether., but only when mice were challenged with sporozoites. The vaccine however, was weakest against direct blood stage challenge, seeing only 76% survival in vaccinated mice. With the exception of contaminated blood, natural challenges will come from sporozoites and AdM42 has demonstrated good efficacy in mice. The promises of AdM42 may however not hold up when translated to human studies given sequence variation in the regional composition of MSP antigens (4, 6), however if like GMZ2 the antigens chosen are highly conserved, this vaccine method may be a promising future malarial candidate.
Transmission Blocking and DNA Based “Altruistic” Vaccines
Nucleic acid based vaccines are a novel concept emerging in 1990 after plasmid transfection of eukaryotic cells was demonstrated (15). DNA vaccines function by expressing pathogenic proteins in host cells, the expressed protein is then presented to adaptive immune cells on MHC molecules (dependent on cell line, this may be MHCI or II) (9). The vaccines are fairly novel, but depending on the gene fragment or product selected, will act in much the same way as the subunit vaccine, but may produce a more long-lasting immunogenic stimulus through prolonged expression in host cells.
“Altruistic” vaccination has been an area of research since 1995 (16) and can broadly be defined as: vaccinating an individual for no clinical benefit, with the intent of blocking transmission (10). The central targets of transmission breaking malaria vaccines are those forms that comprise the sexual stages of P. falciparum’s lifestyle. The sexual stages of malaria are initiated in the vertebrate host, gametocytes form in the human host and fertilisation of oocytes occurs in the invertebrate Anopheles host (4). Targets for transmission blocking may be in the human host, or outside of it. These targets will require different strategies to elicit blocking immunogenicity. As fertilisation occurs outside the human host, mosquito receptors and antigens present post-fertilisation are not displayed to the adaptive immune system and thus do not elicit an immune response. DNA vaccines make this prospect of immunity to targets outside the host feasible. DNA based altruistic vaccines present a steady level of antigen to the immune system, maintaining T and antibody based immunity to the target antigens (16). Upon taking a blood meal, the Anopheles mosquito also ingests the T cells and antibodies directed at either the P. falciparum or Anopheles targets. The effect is the blocking of fertilisation, implantation or migration or total destruction of pre-transmissible particles in the mosquito, preventing further infections. Similarly, fusion proteins against human host sexual stages are able to elicit immune responses capable of binding and killing of many gametocytes, preventing their uptake during a blood meal (8). However, it is unlikely that these vaccines will be funded as there is much interest in measurable clinical outcomes and there is an associated difficulty in measuring to what degree their blocking ability attenuates an outbreak. Whilst these vaccines may not make it to the production phase, they may play a large role in preventing adults with protective immunity from acting as reservoirs of infection.
Considerations For Successful Anti Malaria Vaccines
Distribution of a vaccine is pivotal to a successful immunisation campaign. Distribution requires consideration of geographical and population based factors. Research conducted on RTS,S indicates that the ideal target population for a successfully conducted protective vaccine campaign are infants between 0-2 months (5) ideally any child under 5 years of age should be vaccinated. These infants fall within the most vulnerable age range and further, in endemic areas are likely to encounter infected mosquitoes. Additionally, children’s spleens are not fully developed until around age 2 making it even more difficult for children to detect the blood-borne infection and successfully overcome it. Early immunisation (once a successfully efficacious vaccine is developed) in the youngest members of sub-Saharan communities may prevent most of the P. falciparum fatalities, whilst also preventing progression and transmission of further disease (4, 5, 8). Successful immunity requires the promotion of the survival of effector cells which recognise the specific antigen. Short lived immune responses found in malaria vaccines being developed may require multiple boostings to generate immune memory (4). This makes distributing the vaccine to individuals over a large geographical area with little modern technology and few transport routes, a potentially great obstacle. The maintenance of cold chain would be a large hurdle if the future vaccine required refrigeration (17). Vaccination programmes are present in Africa and it may be necessary to add a future malaria vaccine to the existing schedule and make use of available infrastructure and resources.
Sub-Saharan Africa is fraught with many infectious and non-infectious disease issues. Chief among the infectious diseases, HIV AIDS decimates the immune system of many infected individuals. The damage the virus causes to CD4+ T-cells may damage a pivotal part of the adaptive immune system’s malaria control response (4, 8, 9). Malnutrition is also associated with poor immunity (13). Hunger and famine in many African regions may still pose a barrier to successful immunisation against P. falciparum. Africa is also home to many of humanity’s closest evolutionary ancestors, and it is unknown if they may act as a natural reservoir of malaria, or whether they may seed another P. knowlsei event should a malaria vaccine successfully remove P. falciparum from populations. The goal of reaching a malaria vaccine with 80% efficacy is still a distant prospect, depending on its formulation, whether or not it conforms to the properties of an ideal vaccine, it may be necessary to work around the vaccine’s requirements.
References
- Harrison’s Principles of Internal Medicine, Manual of Medicine. USA: Mc-Graw-Hill Companies; 2009.
- Draper SJ, Goodman AL, Biswas S, Forbes EK, Moore AC, Gilbert SC, et al. Recombinant Viral Vaccines Exposing Merozoite Surface Protein-1 Induce Antibody- and T Cell-Mediated Multistage Protection against Malaria. Cell Host & Microbe. 2004;5:95-105. [PMID 19154991]
- Li S, Et A. Viral vectors for malaria vaccine development. Vaccine. 2007;25:2567-74. [PMID 16914237]
- Hafalla JC, Silvie O, Matuschewski K. Cell biology and immunology of malaria. Immunological Reviews. 2011;240:297-316. [PMID 21349101]
- Asante KP, Et A. Safety and efficacy of the RTS,S/AS01E candidate malaria vaccine given with expanded-programme-on-immunisation vaccines: 19 month follow-up of a randomised, open-label, phase 2 trial. Lancet Infect Dis. 2011;11:1-9. [PMID 21782519]
- Borrmann S, Matuschewski K. Protective immunity against malaria by ‘natural immunization’: a question of dose, parasite diversity, or both? Current Opinion in Immunology. 2011;23:500-8. [PMID 21719266]
- Nduati E, Gwela A, Karanja H, Muygenyi C, Langhorne J, Marsh K, et al. The Plasma Concentration of B Cell Acting Factor is Increased in Children With Acute Malaria. Journal of Infectious DIseases. 2011;204:962-70. [PMID 21849293]
- VandenBerg JP. Reflections on early malaria vaccine studies, the first successful human malaria vaccination, and beyond. Vaccine. 2009;27:2-9. [PMID 18973784]
- Moorthy VS, Good MF, Hill AVS. Malaria vaccine developments. Lancet. 2004;363:150-6. [PMID 14726170]
- Mordmüller B, Et A. Safety and immunogenicity of the malaria vaccine candidate GMZ2 in malaria-exposed, adult individuals from Lambaréné, Gabon. Vaccine. 2010;28:6698-703. [PMID 20696154]
- Mueller A-K, Labaied M, Kappe SHI, Matuschewski K. Genetically modified Plasmodium parasites as a protective experimental malaria vaccine. Nature. 2005;433:163-7. [PMID 15580261]
- Aide P, Et A. Four year immunogenicity of the RTS,S/AS02A malaria vaccine in Mozambican children during a phase IIb trial. Vaccine. 2010;29:6059-67. [PMID 21443960]
- Olotu A, Et A. Efficacy of RTS,S/AS01E malaria vaccine and exploratory analysis on anti-circumsporozoite antibody titres and protection in children aged 5-17 months in Kenya and Tanzania: a randomised control trial. Lancet Infect Dis. 2011;11:103-9. [PMID 21237715]
- Susana Lousada-Dietricha, Et A. A synthetic TLR4 agonist formulated in an emulsion enhances humoral and Type 1 cellular immune responses against GMZ2 – A GLURP– MSP3 fusion protein malaria vaccine candidate. Vaccine. 2011;29:3284-92. [PMID 21349366]
- Liu MA. DNA vaccines: a review. Internal Medicine. 2003;253:402-10. [PMID 12653868]
- Coutinho-Abreu IV, Ramalho-Ortigao M. Transmission blocking vaccines to control insect-borne diseases- A Review. Mem Inst Oswaldo Cruz, Rio de Jeneiro. 2010;105:1-12. [PMID 20209323]
- MVI P. Public-sector preferences for RTS,S/AS01 malaria vaccine formulation, presentation, and packaging. Bathesda: MVI Path2008.

Molecular microbiologist, Post-Doc in infectious diseases research . Research focus on ESBL and carbapenemases. Current UWA Medical student | @CdrHBiscuitIII | LinkedIn |
