
![]()
Palpable Excitement
aka Immunological Inquisition 003
A 76 year-old man presents with a florid spreading violaceous rash over most of his body. He noticed it on his feet when he awoke at 7am and it is now 3pm. His observations are unremarkable.
This is a photograph of his legs:
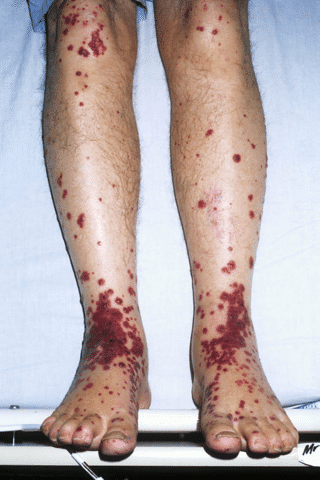
He has no fever, and the rash is slightly itchy.
The day previously he had received intravenous 5FU for Duke’s B stage bowel cancer. He is taking no other medication. He mentions that he has had a dry cough which he has not told his oncologist about.
These are the lesions on his right hand:

Questions
Q1. What is this skin manifestation?
Answer and interpretation
Palpable purpura
Purpura are non blanching violaceous (purple) discolorations of the skin due to blood that has extravasated outside of vessel walls. In this case they are raised and palpable.
Q2. What underlying pathological process does this imply?
Answer and interpretation
Palpable purpura impliesleukocytoclastic vasculitis — a histopathologic term commonly used to denote a small-vessel vasculitis.
It gets its name because ‘broken up white cells’ are seen under the the microscope in pathology specimens. Other causes of purpura, such as thrombocytopenia and pigmented purpuric dermatoses, are not usually palpable.
Stedman’s medical dictionary defines leukocytoclastic vasculitis as:
“Acute cutaneous vasculitis that is characterized by purpura, especially of the legs, and by exudation of neutrophils and sometimes fibrin around dermal venules, with extravasation of red blood cells.”
Leukocytoclastic vasculitis (LCV is, also known as hypersensitivity vasculitis and hypersensitivity angiitis.
Q3. What are the possible underlying causes?
Answer and interpretation
Leukocytoclastic vasculitis may be caused by a number of conditions including:
- drug reactions —
e.g. antibiotics, NSAIDs, thiazide diuretics, serum sickness - infections —
e.g. hepatitis B and C, HIV, endocarditis - malignancy
- autoimmune/ collagen vascular diseases —
e.g. Henoch-Schonlein purpura, granulomatous vasculitides, polyarteritis nodosa, SLE, rheumatoid arthritis and dermatomyositis, among others
Common causes of palpable purpura often can be identified during the patient’s history and physical examination. When this condition occurs in a patient who has not had a known precipitating event, or when the cause is unclear from the history and physical examination, diagnosis may be based on findings of the laboratory investigations and skin biopsy.
Many cases of palpable purpura are idiopathic and self-limited, with no cause being found about 50% of the time. When treatment is necessary, dapsone, colchicine or systemic corticosteroids are useful drugs.
Q4. What is the most important cause to exclude and/or treat in the emergency setting?
Answer and interpretation
Sepsis — especially meningococcemia
This patient was given was given ceftriaxone 2g IV immediately after 2 sets of blood cultures were taken.
You assess the patient further to try to elucidate the cause of his rash:
Infection seemed unlikely given that he looked and felt well. His CBC shows a slight neutrophilia and his CRP was elevated at 75. His cultures grew nothing after five days.
Other infections that can involved in immune complex production are viral (Hepatitis C, B and HIV) and intravascular infections such as endocarditis. On your clinical information system you can see recent negative Hepatitis B and C and HIV serologies. There is no murmur on examination and he has no history of valvular heart disease. An echocardiogram last week ordered by his oncologist was negative.
Drugs as a cause of hypersensitivity vasculitis are most often antibiotics principally penicillins and cephalosporins. Serum sickness, initially due to horse derived anti-sera, may also have a vasculitic component. He has had none of these recently.
A chest radiograph is obtained and shows an ill-defined mass in the right upper lobe. The patient is transferred to the ward with the possibility of metastatic disease being be entertained, although primary immunologic disease still needed to be excluded.
Q5. Could the patient have Henoch-Schönlein Purpura?
Answer and interpretation
Henoch-Schonlein purpura (IgA seen on skin biopsy in the vessels by immunofluorescence) can present like this.
Usually patients have abdominal pain, though the presentation in adults can involve many tissues/organs and can be life threatening. If a biopsy is taken, it is critical a new lesion is biopsied as IgA has a short life span in the vessel walls. Samples should be taken for Immunofluorescence (not in formalin) and another for histopathology.
Whilst on the ward, a skin biopsy revealed classic leucocytoclastic vasculitis without immune deposition.
Q6. Could the patient have an ANCA-associated vasculitis?
Answer and interpretation
ANCA (anti-neutrophil cytoplasmic antibody) associated vasculitis can occasionally present like this.
In addition, in drug-induced ANCA disease, prominent skin involvement occasionally with necrosis can be seen; offending drugs include but are not limited to propylthiouracil, Carbimazole and hydralazine. Drug cessation and treatment is needed; in some the disease continues despite drug cessation.
A day later his ANCA results return.
Of note, it is possible to run these tests urgently, even after hours, though given the expense of this for the laboratory, should only be asked for in life threatening situations (pulmonary haemorrhage, acute renal failure).
His ANCA results reveal the following pattern:
- The cells are fixed neutrophils and show a classic cytoplasmic (C-ANCA) with perinuclear accentuation and internuclear accentuation.
- A second phase test for proteinase-3 antibodies was strongly positive.

These test results are usually seen in Granulomatosis with polyangiitis (Wegener’s granulomatosis) though occasionally can be seen in endocarditis.
The lung lesion was biopsied and showed non-caseating granulomatous inflammation which together with the other pathology was diagnostic of…
Reveal the diagnosis!
Granulomatosis with polyangiitis (GPA) (Wegener granulomatosis)
The eponym ‘Wegener Granulomatosis’ has been replaced with the term ‘Granulomatosis with polyangiitis (GPA)‘. This transition was in part to achieve nomenclature symmetry with Microscopic Polyangiitis (MPA) and Eosinophilic granulomatosis with polyangiitis (EGPA, Churg-Strauss); and to remove any historical association with Friedrich Wegener following revelations of his association with the Nazi Party in World War II
Granulomatosis with polyangiitis (GPA) is a rare multisystem autoimmune disease of unknown etiology. Its hallmark features include necrotizing granulomatous inflammation and pauci-immune vasculitis in small- and medium-sized blood vessels.
Clinical features include:
- nonspecific consitutional symptoms, e.g. poor appetie, weight loss, fatigue, fevers
- ocular —
e.g. conjunctivits, episcleritis, uveitis, optic nerve vasculitis, retinal vessel occlusion, proptosis - ENT —
e.g. sinusitis, rhinitis, epistaxis, otis media, tracheal and subglottic granulomas - Pulmonary —
e.g. cough, lung infiltrates, hemoptysis, diffuse alveolar hemorrhage - musculoskeltal involvement
e.g. polyarticular symmetrical arthralgias +/- arthritis - Renal —
e.g. crescentic necrotizing glomerulonephritis, renal failure - Neurological —
e.g. mononeuritis multiplex, sensorimotor polyneuropathy,cranial nerve palsies, CNS vasculitis and granulomatous lesions. - Skin —
e.g. palpable purpura, skin ulcers.
This patient in this case was started on steroids and methotrexate and responded well, last seen by the author 1 year after diagnosis.

CLINICAL CASES
Immunological Inquisition
BA MA (Oxon) MBChB (Edin) FACEM FFSEM. Emergency physician, Sir Charles Gairdner Hospital. Passion for rugby; medical history; medical education; and asynchronous learning #FOAMed evangelist. Co-founder and CTO of Life in the Fast lane | On Call: Principles and Protocol 4e| Eponyms | Books |