
![]()
Short QT Syndrome
Overview of SQTS
- Familial arrhythmogenic disease associated with paroxysmal atrial and ventricular fibrillation, syncope, and sudden cardiac death
- Genetically-inherited cardiac channelopathy, on the same spectrum as other familial arrhythmogenic diseases such as Long QT syndrome (LQTS), Brugada Syndrome, and Catecholamine Polymorphic Ventricular Tachycardia (CPVT)
- Patients are typically young and healthy, with no structural heart abnormalities; age at first presentation ranges from a few months to the sixth decade of life (median age = 30 years)
- The most common initial presenting symptom is cardiac arrest (in one-third of cases); other patients may present with palpitations or syncope due to rapid atrial fibrillation or self-terminating ventricular arrhythmias
- Witnessed cardiac arrest within the first year of life and unexplained infant deaths have been observed in patients and families with SQTS, making it a possible cause of sudden infant death syndrome (SIDS)
- SQTS is still a relatively new disease – it was first described in 2000, and elucidation of the genetic, electrophysiological and clinical abnormalities associated with the disease has only taken place over the past few years
Diagnosis
At present, there are no diagnostic criteria for SQTS. The diagnosis is based upon the patient’s symptoms (e.g. syncope, palpitations), family history (of syncope, sudden death or atrial fibrillation at an early age) and characteristic findings on the 12-lead ECG.
Clinical features
So far, there have only been a handful of cases of SQTS reported in the literature. The true prevalence of the disease is unknown. The largest case series to date reported on 29 patients with the disease:
- The most common presenting symptom was cardiac arrest (in one-third of cases)
- Cardiac arrest occurred in the first months of life in two patients.
- Syncope was the presenting symptom in 24% of cases, thought to be secondary to self-terminating episodes of ventricular fibrillation
- Up to 31% of patients complained of palpitations, and 80% of patients had documented episodes ofatrial fibrillation
- All patients had a QT < 320ms and a QTc < 340ms with no evidence of structural heart disease(NB. This case series did not include the more recently-described SQTS genotypes 4 & 5)
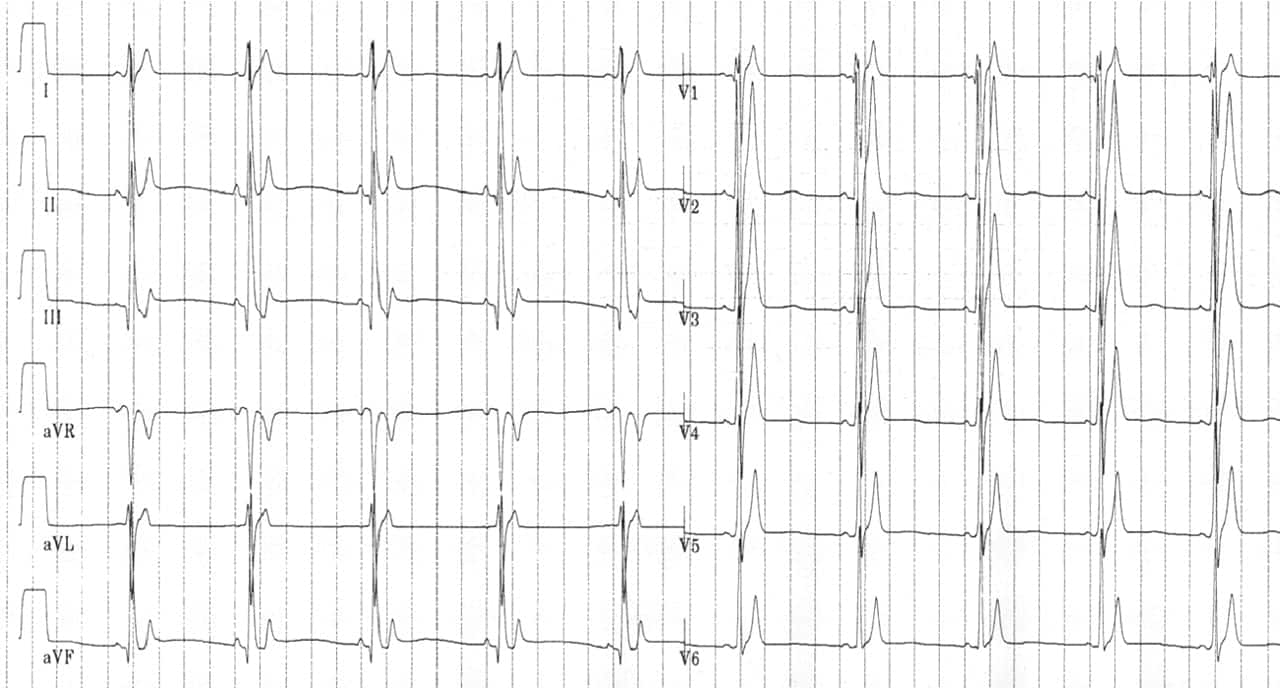
ECG features
- Short QT interval
- Lack of normal changes in QT interval with heart rate
- Peaked T waves, particularly in precordial leads
- Short, or absent, ST segments
- Paroxysmal episodes of atrial or ventricular fibrillation
QT, ST and T-wave changes in SQTS
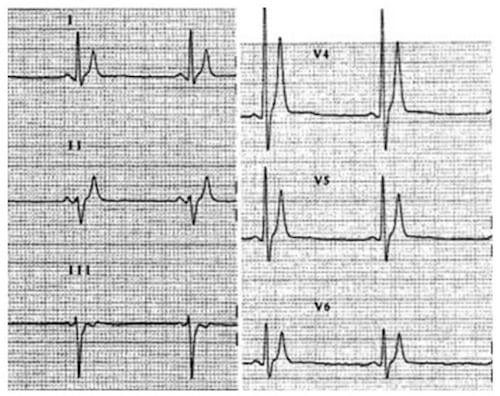
Short QT interval, peaked T waves and short ST segments in two patients with SQTS 1.
Mechanism
Arrhythmogenesis in SQTS is thought to result from:
- Extremely short atrial and ventricular refractory periods (manifest on the ECG as a short QT interval)
- Transmural dispersion of repolarisation, i.e., the different layers of the myocardium (endocardium, epicardium and the mid-myocardial ‘M-cells’) repolarise at different rates
Both these repolarisation abnormalities convey an increased susceptibility to re-entrant atrial and ventricular arrhythmias.
Short QT interval
There is currently no universally accepted lower limit of normal for the QT interval that can be used to diagnose SQTS.
- Known patients with SQTS genotypes 1-3 all had QTc intervals < 300-320 ms
- Known patients with SQTS genotypes 4 & 5 all had QTc intervals < 360 ms
A recent review by Viskin suggested the following approach:
- QTc intervals < 330 ms in males or < 340 ms in females should be considered diagnostic of SQTS
- QTc intervals < 360 ms in males or < 370 ms in females should only be considered diagnostic of SQTS when supported by symptoms or family history

Lack of normal changes in QT with heart rate
- Patients with SQTS demonstrate fixed QT intervals which remain constant over a range of heart rates
- At fast heart rates, the calculated QTc may appear normal (= ”pseudonormal” QTc)
- However, as the heart rate slows, the QTc typically fails to prolong.
- Serial ECGs or Holter monitoring at rest may be used to try and capture short QT intervals during periods of relative bradycardia (heart rate 60-80bpm)
- Exercise testing may demonstrate lack of adaptation of QT interval with different heart rates.
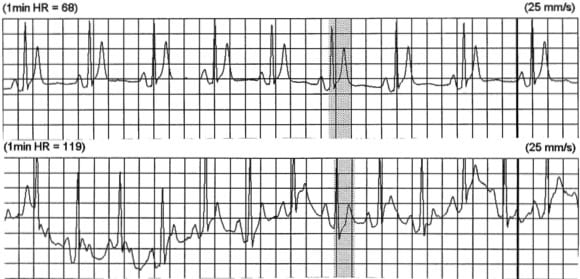
- Holter strip from a patient with SQTS at heart rates of 68 and 119 bpm.
- QT interval of 280 ms remains constant at both heart rates.
- Reproduced from Short QT Syndrome.org
Electrophysiological Studies
Electrophysiological studies in SQTS demonstrate:
- Extremely short atrial and ventricular refractory periods
- High rates of inducible atrial and ventricular fibrillation
- Marked vulnerability to mechanical induction of ventricular fibrillation
The role of EP studies in diagnosing and risk-stratifying patients with SQTS has not yet been established
Treatment Options
- At present, the only effective treatment is implantation of an ICD
- The main problem with this is T-wave oversensing and inappropriate shocks due to the tall, narrow T waves seen in SQTS
- Efforts to find a suitable pharmacological treatment have focused on potassium blocking anti-arrhythmic agents (classes Ia and III)
- Class III agents ibutilide and sotalol, while having theoretical benefits in prolonging QT and suppressing arrhythmias, have been shown to be ineffective due to reduced drug binding to mutated potassium channels
- Class Ia agents quinidine and disopyramide have shown more promising effects. Quinidine is currently the agent of choice, having been shown in SQTS 1 patients to markedly prolong both the QT interval and ventricular refractory period, with normalisation of ST segments and T waves and prevention of VF induction
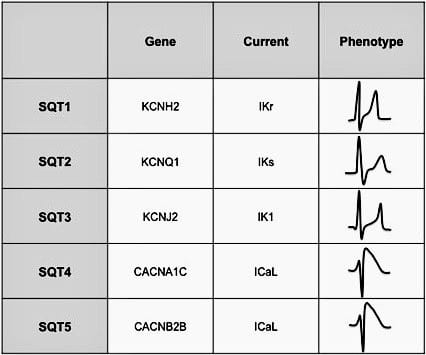
Genetic Basis
- SQTS is a genetically heterogenous disease, with multiple mutations producing a similar clinical picture. Five mutations have been characterised so far, all of which seem to be inherited in an autosomal dominant fashion.
- SQTS genotypes 1-3 are produced by a gain-of-function mutation in myocardial potassium channels (the opposite to LQTS), with increased potassium efflux during various stages of the action potential leading to more rapid atrial and ventricular repolarisation with marked shortening of the QT interval (<320 ms).
- SQTS genotypes 4 and 5 are produced by a loss-of-function mutation in the L-type cardiac channel, with reduced influx of calcium during the plateau phase of the action potential leading to modest shortening of the QT interval (<360ms) associated with a Brugada-syndrome-like QRS complex morphology.
Potassium fluxes in SQTS 1-3
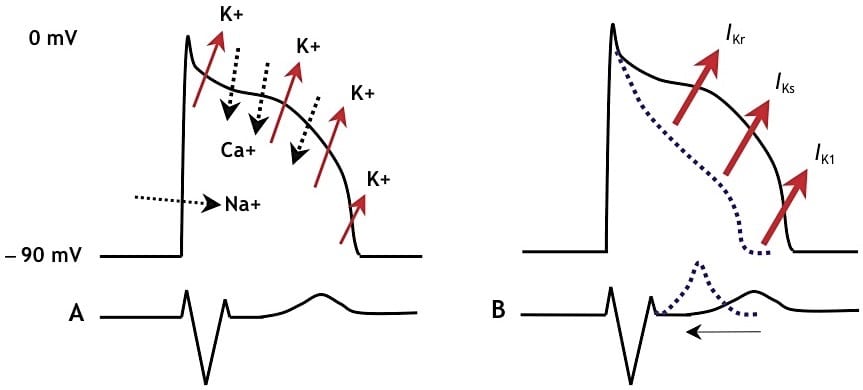
- A. Schematic representation of the normal action potential and the flux of ions
- B. With gain-of-function mutations in any of 3 different potassium channels (SQTS 1-3), the cardiac action potential shortens and the QT interval decreases
Classification of SQTS according to genotype
Related Topics
References
- Gaita F, et al. Short QT Syndrome: a familial cause of sudden death. Circulation. 2003 Aug 26;108(8):965-70. Epub 2003
- Brugada R, Hong K, Cordeiro JM, Dumaine R. Short QT syndrome. CMAJ. 2005 Nov 22;173(11):1349-54.
- Schimpf R, et al. Short QT syndrome. Cardiovasc Res. 2005 Aug 15;67(3):357-66
- Giustetto C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J. 2006 Oct;27(20):2440-7.
- Viskin S. The QT interval: too long, too short or just right. Heart Rhythm. 2009 May;6(5):711-5. Epub 2009
- Crotti L, Taravelli E, Girardengo G, Schwartz PJ. Congenital short QT syndrome. Indian Pacing Electrophysiol J. 2010 Feb 1; 10(2):86-95
- Moreno-Reviriego S, Merino JL. Short QT syndrome. An article from the E-Journal of the ESC Council for Cardiology Practice.
- Short QT Syndrome Online resource
Advanced Reading
Online
- Wiesbauer F, Kühn P. ECG Mastery: Yellow Belt online course. Understand ECG basics. Medmastery
- Wiesbauer F, Kühn P. ECG Mastery: Blue Belt online course: Become an ECG expert. Medmastery
- Kühn P, Houghton A. ECG Mastery: Black Belt Workshop. Advanced ECG interpretation. Medmastery
- Rawshani A. Clinical ECG Interpretation ECG Waves
- Smith SW. Dr Smith’s ECG blog.
- Wiesbauer F. Little Black Book of ECG Secrets. Medmastery PDF
Textbooks
- Zimmerman FH. ECG Core Curriculum. 2023
- Mattu A, Berberian J, Brady WJ. Emergency ECGs: Case-Based Review and Interpretations, 2022
- Straus DG, Schocken DD. Marriott’s Practical Electrocardiography 13e, 2021
- Brady WJ, Lipinski MJ et al. Electrocardiogram in Clinical Medicine. 1e, 2020
- Mattu A, Tabas JA, Brady WJ. Electrocardiography in Emergency, Acute, and Critical Care. 2e, 2019
- Hampton J, Adlam D. The ECG Made Practical 7e, 2019
- Kühn P, Lang C, Wiesbauer F. ECG Mastery: The Simplest Way to Learn the ECG. 2015
- Grauer K. ECG Pocket Brain (Expanded) 6e, 2014
- Surawicz B, Knilans T. Chou’s Electrocardiography in Clinical Practice: Adult and Pediatric 6e, 2008
- Chan TC. ECG in Emergency Medicine and Acute Care 1e, 2004
LITFL Further Reading
- ECG Library Basics – Waves, Intervals, Segments and Clinical Interpretation
- ECG A to Z by diagnosis – ECG interpretation in clinical context
- ECG Exigency and Cardiovascular Curveball – ECG Clinical Cases
- 100 ECG Quiz – Self-assessment tool for examination practice
- ECG Reference SITES and BOOKS – the best of the rest
ECG LIBRARY
Emergency Physician in Prehospital and Retrieval Medicine in Sydney, Australia. He has a passion for ECG interpretation and medical education | ECG Library |

MBBS FACEM DDU (Emergency) CCPU. Emergency Physician in Melbourne, Australia. Co-Ultrasound Lead for Emergency Medicine at The Alfred Hospital. Special interests in diagnostic and procedural ultrasound, medical education, and ECG interpretation. Editor of the LITFL ECG Library.
Of note is, that there have only a few families around the world (!) been identified with congenital short QT-Syndrome. Hence this is a crazy rarity not really worth remembering much about. There are certainly more important ECG things to know.